Antibody Optimization¶
Harness the power of advanced AI models to evaluate, screen, and optimize your lead antibodies efficiently.
If you have a single lead antibody, you can humanize it in one click and perform virtual screening on the generated sequences to select the best candidates for experimental validation.
If you have multiple lead antibodies, our task allows comprehensive scoring based on humanness, surface charge, hydrophobicity, isoelectric point, expression levels, structural confidence, and PTM risks. You can rank and screen molecules based on these scores, with intuitive color-coded visualizations at the amino acid level displayed on both sequence and structure.
If you need to modify existing antibodies for better affinity, stability and developability, the Antibody Mutation Advisor provides intelligent mutation recommendations to guide your antibody engineering efforts.
Inputs¶
To submit an Antibody Optimization job, open the Project Editor and click "New Job" button on the left sidebar. Then click "Antibody Optimization" under the "Antibody Design" group to open the job submission page.
- Sequence type: Type of the antibodies to be optimized. Options include "VH+VL" (default) and "VH".
- Antibodies: Input one or more antibodies for optimization, evaluation, or screening. Enter sequences directly, upload a FASTA file (click the "
 " button), or paste the content of a FASTA file into the input box.
" button), or paste the content of a FASTA file into the input box.- Antibody Name: Defaults to "Antibody". Hover over the name and click "
 " to change it.
" to change it. - Sequence: Sequences of the VH/VL regions of each antibody.
- : Upload a .FASTA file with two chains labeled with the same prefix ending in "_H" and "_L". A valid example is shown above.
- Antibody Name: Defaults to "Antibody". Hover over the name and click "
- Job Name: The name of the job. Note that the job name must be unique within the project.

Parameters¶
The parameters for this task include:
- Scheme: The protocol to number each antibody's amino acids, aligning equivalent amino acids across different antibodies. Options include "Kabat", "IMGT", "Chothia", or "AHo".
- CDR def.: The definition of CDR regions, affecting CDR grafting and one-click humanization results. Options include "Kabat", "IMGT", "Chothia", or "North". Here we compare different CDR definitions. We recommend using "North" for one-click humanization.
- Species: Species to search for VH/VL germlines. Options include "human" (default), "cat", "mouse", "rat", "rabbit", "alpaca", "pig", and "rhesus".
-
Mutator: The protocol to mutate the starting sequence. Only applicable when the input sequence has only one sequence. Options include:
Mutator
No mutation. The result table will show the input antibodies as the initial outputs.
The traditional humanization technique that grafts the CDR regions of the input antibody onto human germline sequences. The result table will show one humanized sequence apart from the input sequence. Below are the parameters for this mutator.
- VH germline: Germline heavy V gene to use as template for CDR grafting. Defaults to 'Auto', which selects the germline closest to the parental sequence for the chosen species.
- VL germline: Germline light V gene to use as template for CDR grafting. Defaults to 'Auto', which selects the germline closest to the parental sequence for the chosen species.
- Keep Vernier: Whether to keep Vernier regions as in parental sequence to preserve binding.
- Keep VH-VL interface: Whether to keep VH-VL interface in parental sequence to preserve antibody stability. Only applicable if "sequence type" is set to "VH+VL".
- Keep Nb KeyRes: Whether to keep nanobody key residues in parental sequence to preserve solubility and stability. Only applicable if "sequence type" is set to "VH".
The proprietary humanization algorithm developed by BioGeometry, which transforms the antibody into a more human-like molecule with fewer mutations. The result table will show one humanized sequence apart from the input sequence. Below are the parameters for this mutator.
- # Iterations: Number of humanization iterations for the GeoHumAb algorithm. Defaults to 1. Higher value will result in better humanization but worse sequence preservation.
- Keep CDRs: Whether to keep CDR regions as in parental sequence to preserve binding.
- Keep Vernier: Whether to keep Vernier regions as in parental sequence to preserve binding.
- Keep VH-VL interface: Whether to keep VH-VL interface in parental sequence to preserve antibody stability. Only applicable if "sequence type" is set to "VH+VL".
- Keep Nb KeyRes: Whether to keep nanobody key residues in parental sequence to preserve solubility and stability. Only applicable if "sequence type" is set to "VH".
Only applicable if "species" is set to "human".
The proprietary one-click humanization algorithm developed by BioGeometry, which combines VH-VL pairing, CDR grafting, selective backmutation, and obtains multiple humanized sequences for subsequent calculations. The result table will show multiple humanized sequences apart from the input sequence. Below are the parameters for this mutator.
- VH germline: Germline heavy V gene to use as template for CDR grafting. Defaults to 'Auto', which selects the human germline closest to the parental sequence.
- VL germline: Germline light V gene to use as template for CDR grafting. Defaults to 'Auto', which selects the human germline closest to the parental sequence.
Only applicable if "species" is set to "human".
Results¶
In the File & Job Manager panel, click the name of the job you previously created to view the job results.

The results are presented in three pages:
- Summary Page: where the properties for all antibodies are listed. New antibody sequences and new property metrics can be added from this page.
- Compare Page: where the sequences and per-residue properties of selected antibodies are compared along with the overall properties.
- Edit Page: where you can optimize a specific antibody sequence with an AI assistant.
Summary Page¶
In the Antibody Properties table in the summary page (shown above), the name, VH ID, VL ID (if the sequence type is "VH+VL"), and predicted properties of all antibody molecules in the task are listed.
The antibodies may either come from job inputs, by a mutation algorithm, or by manual editing on the Edit Page. The properties listed include:
Antibody Properties
- Name: The name of the antibody. You can rename an antibody by clicking the "" button in its "name" column. Note that the name must be unique within the job.
- VH ID: The identifier of the heavy chain sequence. You can rename it by clicking the "" button in its "VH ID" column. Note that all antibodies with the same VH sequences will be renamed together.
- VL ID: The ID of the light chain of the antibody. You can rename it by clicking the "" button in its "VL ID" column. Note that all antibodies with the same VL sequences will be renamed together.
- Humanness (percentile): The humanness score of the antibody and its percentile in our antibody library. Generally, antibodies with a percentile below 10% are considered to have a high immunogenicity risk.
- Germline content: The percentage of amino acids in the antibody that are identical to the germline sequences. Generally, it should be greater than 80%.
- FR Germline content: The percentage of amino acids in the FR region of the antibody that are identical to the germline sequences. Generally, it should be greater than 85%.
- Germline allele: The germline genes of the antibody. The heavy and light chains are separated by "|", such as "IGHV1-2*02 | IGKV4-1*01".
- Optimal pH: The pH value at which the antibody structure is most stable.
- pI: The isoelectric point of the antibody calculated based on the structure.
- Positive patch (percentile): The number of positive charge patches on the antibody surface and its percentile. A percentile greater than 90% indicates a high risk of aggregation.
- Negative patch (percentile): The number of negative charge patches on the antibody surface and its percentile. A percentile greater than 90% indicates a high risk of aggregation.
- Charge symmetry (percentile): The charge symmetry of the VH and VL regions (the product of the net surface charge of amino acids). A percentile less than 10% indicates a high risk of aggregation.
- Hydrophobic patch (percentile): The number of hydrophobic patches on the antibody surface. A percentile less than 10% or greater than 90% indicates a high risk of aggregation.
- Thermostability (percentile): The thermostability of the antibody and its percentile. A percentile less than 10% indicates a high risk of thermostability. Currently only available for "VH" sequence type (i.e. nanobodies).
- Expression (percentile): The expression level in CHO cells of the antibody and its percentile in our antibody library. A percentile less than 10% indicates a high risk of low expression. Currently only available for "VH+VL" sequence type.
- Purity (percentile): The purity of the antibody after one step of A-column purification (detected by SEC-HPLC). A percentile less than 10% indicates a high risk of low purity. Currently only available for "VH+VL" sequence type.
- #PTMs: The number of translation post-translational modifications (PTMs) and non-specific binding risk sites on the antibody.
- #PTMs (CDR/Vernier): The number of PTMs and non-specific binding risk sites on the CDR and Vernier regions of the antibody. Hover on the number to view the detailed risk sites.
- Energy (Amber14SB): The energy of the antibody structure calculated using the Amber14SB force field.
- plDDT: The local distance difference test (lDDT) of the entire antibody, ranging from 0 to 100. The higher the value, the more confident the antibody structure prediction. It has a positive correlation with antibody structural stability.
- CDR plDDT: The lDDT of the CDR region of the antibody, ranging from 0 to 100. The higher the value, the more confident the antibody CDR structure prediction.
- wpTm: The weighted TM score of the antibody VH-VL complex, ranging from 0 to 1. The higher the value, the more confident the antibody VH-VL complex structure prediction. It has a positive correlation with the VH-VL pairing propensity.
- RMSD to {{ref}}: The root mean square deviation (RMSD) of the antibody structure compared to the reference structure.
- CDR RMSD to {{ref}}: The RMSD of the CDR region of the antibody compared to the reference CDR region. Lower values indicate better CDR structure preservation and usually, higher binding affinity.
- ΔFitness (heavy/light) to {{ref}}: The change in fitness of the heavy/light chain compared to the reference sequence. Higher values indicate better fitness.
The above columns will not appear by default. You can click "![]() " in the table header and specify the reference sequence and the placeholder content of "{{ref}}" in the column name to display these columns.
" in the table header and specify the reference sequence and the placeholder content of "{{ref}}" in the column name to display these columns.
In the header of each property column, there are two small buttons you can press:
 Sort: Click the up/down triangles to sort the current column in ascending/descending order, respectively.
Sort: Click the up/down triangles to sort the current column in ascending/descending order, respectively. Filter: Click to filter the numerical values in the current column.
Filter: Click to filter the numerical values in the current column.
Above the table are five buttons to perform various operations:
| Icon | Operation | Description |
|---|---|---|
| Customize Columns | Customize which properties to display and their order. You might want to focus on a few columns and hide the rest. | |
| Batch Operation | Click to perform batch operations on multiple antibodies (rows) in the table. Supported operations include:
|
|
| Score mutants | Add columns to show the structure similarity/fitness change scores of all antibodies as mutants of some reference antibody. | |
| Add Sequences | Click to add new sequences to the table. After clicking the button, input the new sequences in the pop-up window. | |
| Compare Sequences | Align and compare the sequences of the selected antibodies. Click to go to the Compare Page, which shows the molecule-level and amino acid-level scores of each antibody. |
Compare Page¶
This page (titled “Antibody Property Comparison”) will align the selected sequences and compare their molecule-level and residue-level scores.

Click the job name in parentheses at the top of the page ("hSM_opt" in the above figure) to return to the Summary Page.

This page consists of two sections: "Antibody Properties" which displays molecule-level scores and "Per-residue scores" which displays residue-level scores.
Antibody properties¶
In the "Antibody Properties" section, the properties of the selected antibodies are listed as a table. As in the Result Table, you can click the ![]() button to customize which properties to display and their order, or click the
button to customize which properties to display and their order, or click the ![]() button to perform batch operations on the selected antibodies, such as copying the FASTA sequences or exporting the CSV table.
button to perform batch operations on the selected antibodies, such as copying the FASTA sequences or exporting the CSV table.
Per-residue scores¶
In the "Per-residue scores" section, the selected antibody sequences are aligned and preseted with a variety of predicted properties (scores). You can change the residue scorer by switching to the corresponding tab.
The background color of each residue represents a certain model's score for that residue. Available models and their coloring schemes are as follows:
Amino Acid Scorers and Coloring Scheme
Shows the amino acid type.
![]()
Shows the predicted risk of inducing immune responses in humans. No risk is white; the deeper the red, the higher the risk.
More specifically, a 9-peptide segment is viewed as risky if it occurs in less than 10% of human beings in our antibody sequence repertoire. The more 9-peptide segments an amino acid belongs to, the deeper the red color.
If this model is enabled in an Edit Page, residues in the current sequence will be highlighted with a red triangle ![]() if it has immunogenicity risk, i.e., has a frequency below 1% in the germline family (e.g. IGHV1). A green triangle
if it has immunogenicity risk, i.e., has a frequency below 1% in the germline family (e.g. IGHV1). A green triangle ![]() indicates that the residue was immunogenicitically risky before editing, but is now not risky after editing.
indicates that the residue was immunogenicitically risky before editing, but is now not risky after editing.
![]()
Shows the predicted pKa of the side chain of each amino acid. Blue indicates a higher pKa (weaker acidic), while red indicates a lower pKa (stronger acidic).
![]()
Shows the predicted net surface charge of each amino acid. Positive charges are blue, while negative charges are red.
![]()
Shows the size of the charged patches on the antibody surface. The larger the patch, the higher the risk of charge-induced aggregation.
![]()
Shows the hydrophobicity of each amino acid. Yellow indicates higher hydrophobicity, while blue indicates lower hydrophobicity.
![]()
Shows the size of the hydrophobic patches on the antibody surface. The larger the patch, the higher the risk of hydrophobicity-induced aggregation.
![]()
Shows the predicted risk of translation post-translational modifications (PTMs) and non-specific binding risk sites on the antibody. The higher the risk, the darker the color.
![]()
In addition to background colors, the following sequence annotations are available:
- CDR regions are underlined in dark gray. Additionally, when applicable, Vernier zones, VH-VL interface regions, and nanobody key residues are highlighted in light gray. These residues are likely to be important for antibody stability and binding affinity. Please be cautious when changing them.
- If you enable "Show solvent accessibility" in the settings, the solvent accessibility of each amino acid is indicated by its font weight and text color: black, bolded residues are exposed to the solvent, while gray, light residues are buried.
- Only the reference sequence is displayed in full, with each amino acid shown as an uppercase letter. In all other sequences, positions where the amino acid is the same as in the reference sequence are represented by a "·" (dot), and positions where the amino acid is different are shown as an uppercase letter. You can change the reference sequence by clicking on the antibody names or VH/VL IDs to the right of the sequences.
- Some sequences may not have a residue in certain positions. In this case, the position is displayed as "-" (dash).
- When you hover over a residue, an info overlay will appear, which displays the position number, region, and score value of that residue. For example: "H113 (IMGT) | CDR3 | score: 0.889". If the scorer is PTM and the residue is risky, the overlay will also display the risk detail. "freq" is the frequency of this risk at the current position; "rsa" is the relative solvent accessibility of the current residue.

Above the table are four buttons to perform various operations:
| Icon | Operation | Description |
|---|---|---|
| Add Sequence | Add new sequences to the comparison. The new sequences may either come from the current job, or from a germline search. | |
| Show All/Unique Sequences | Toggle between showing all selected sequences (identified by antibody name) or only unique sequences (identified by VH/VL ID). | |
| Toggle Chain | Switch between displaying the heavy chain or light chain. | |
| Setting | Open the settings window, where you can modify the following configuration items:
|
Edit Page¶
This page (titled “Editing {antibody_name}”) allows you to edit a specific antibody sequence with AI assistance, generating molecules with multiple properties improved and saving them to the Result Table.

Click the job name in parentheses at the top of the page ("hSM_opt" in the above figure) to return to the Summary Page.

This page consists of the following sections:
- Antibody Properties: displays molecule-level scores for pre-edit and current antibodies
- Per-residue scores: displays residue-level scores
- Mutation advisor: displays the mutations recommended by the AI assistant
- Structure viewer: displays the structure of the current antibody
The first two sections is almost identical to the compare page. The main differences are:
- You can save your current sequence to the result table by clicking the
 button to the right of "Per-residue scores". After saving, the current sequence will remain in your on your page as a reference sequence。
button to the right of "Per-residue scores". After saving, the current sequence will remain in your on your page as a reference sequence。 - You can customize the mutation advisor options by clicking the
 button.
button. - The hover overlay in the per-residue scores section contains mutation advisor results.

Mutation advisor¶
The Mutation advisor provides intelligent mutation recommendations to guide your antibody engineering efforts. You can click the ![]() button to open the settings window and configure the following "Mutation advisor" options:
button to open the settings window and configure the following "Mutation advisor" options:
Mutation advisor options

An AI assistant will provide up to 5 mutation suggestions for each site based on the current antibody sequence. In the default "Raw score" mode, mutations are colored using a red-white-blue gradient scale:
- Red: Mutations predicted to perform worse than wild-type (negative scores)
- White: Neutral mutations (score ≈ 0)
- Blue: Mutations predicted to perform better than wild-type (positive scores)
The color intensity scales differently for positive and negative scores, with different rates of color change per unit of score to optimize visual discrimination.
To make the suggestions clearer and more readable, the current residues in the antibody sequence are represented by dots ("·"); furthermore, residues with confidence below 1% are omitted. For detailed documentation, please refer to the Mutation Advisor User Manual.
The available assistant models (Copilot model) include:
- SaProt: A protein language model enhanced with structure-aware vocabulary. Recommended for optimizing antibodies.
- ESM1v-650M: A language model specifically designed to predict the impact of mutations.
- ESM2-3B: An updated and more powerful general protein language model.
- GeoHumAb: Our proprietary algorithm for humanizing antibodies.
![]()

Displays up to five germline sequences most similar to the current antibody sequence. The residues that agree with the current sequence are highlighted in light blue. To reduce the immunogenicity of the current sequence, you might want to mutate the current residues to the corresponding residues in the germline sequence.
You can customize the source species of the germline sequence in the settings (see below). The available options include: Human (default), Cat, Mouse, Rat, Rabbit, Alpaca, Pig, Rhesus.
![]()

When you hover over a mutation suggestion, the detailed information of the mutation suggestion will appear next to your cursor. It includes:
- Position: The number of the amino acid in the antibody sequence, formatted as "{chain type}{residue number}{insertion code}", e.g., "H52A".
- Region: The antibody segment to which the residue belongs, with possible values of FR1-4 or CDR1-3. The Vernier zones, VH-VL interface regions and nanobody key residues are also marked. These residues are likely to be important for antibody stability and binding affinity. Please be cautious when changing them.
- Advisor confidence: The top 10 mutation suggestions and their predicted likelihoods, with the hovered residue highlighted in blue. The higher the predicted likelihood, the more likely the mutation is to improve the antibody properties (humanness/fitness).
- Risk detail: If the scorer is PTM and the residue is risky, the risk detail will be displayed. "freq" is the frequency of this risk at the current position; "rsa" is the relative solvent accessibility of the current residue.
Below are ways to edit the current sequence. When you have updated the current sequence, you can click the ![]() button to save the current sequence to the result table.
button to save the current sequence to the result table.
Ways to edit the current sequence
Hover over the "Current (mt)" sequence and select "Manually edit sequence" to enter the sequence editing mode. At this point, an editable sequence will appear below the "Current (mt)" sequence, where you can:
- Click the position you want to mutate, and enter the amino acid you want to mutate. Press the Tab or Shift+Tab keys to move left or right between amino acids.
- Click the leftmost position, and press Ctrl+V to paste the mutant sequence.
- Click any amino acid in the per-residue scores or mutation suggestion sections, and the current residue will be mutated to that amino acid.
When you are done editing, click the OK button on the right to update the current sequence.

Hover over the "Current (mt)" sequence and select "Humanize sequence" to open the humanization popup.
Then, you may set the humanization parameters and click "Confirm" to trigger the humanization calculation.
After the calculation is complete, the humanized sequence will be displayed as an editable sequence below the "Current (mt)" sequence, where you can further edit the sequence.
When you are done editing, click the OK button on the right to update the current sequence.

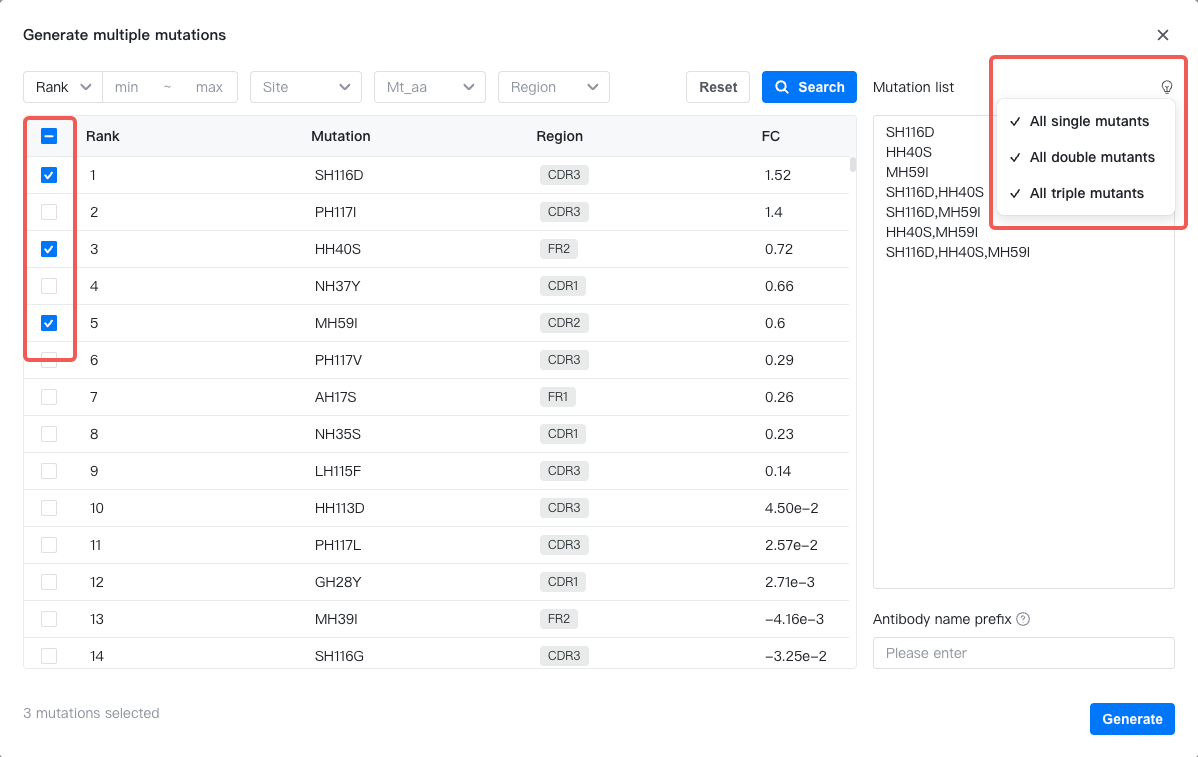
Hover over the "Current (mt)" sequence and select "Generate multiple mutants" to open the mutation generatoin popup.
On the left side of the popup, the AI-suggested mutations are listed in a table. You may use the filter functions to filter the displayed mutations. Select mutations to apply, then click the "![]() " button on the right to generate all single, double and triple mutants comprised of these mutations. You may also manually input the mutation list.
" button on the right to generate all single, double and triple mutants comprised of these mutations. You may also manually input the mutation list.
After you have prepared the mutation list, enter the name prefix for the new antibodies and click "Generate" to add the new antibodies to the result table.
Structure viewer¶
The structure viewer displays the structure of the current antibody.
The amino acid residues in the structure are colored according to the same scheme as the per-residue scores.
Acknowledgements¶
This task is supported by the following models:
- Our proprietary antibody structure prediction model,
- Our proprietary humanization and humanness prediction models,
- Our proprietary expression level/aggregation/thermostability prediction model,
- ESM series models from Meta AI, used for modeling and optimizing protein sequences,
- SaProt from Westlake University, used for modeling and optimizing protein sequences,
- The therapeutic antibody profiler (TAP) model proposed by Oxford University and improved by us, for predicting antibody side chain pKa, surface charge, hydrophobicity, CDR length, etc., and comparing them with clinical-stage antibodies.
- Our proprietary sequence risk analysis model, for predicting post-translational modifications (PTMs) and non-specific binding risk sites.