Targeted CDR Library Design¶
Targeted CDR Library Design is a task to design an antibody CDR library de novo targeting a given epitope. It is especially useful when you want to design antibodies that target a specific epitope that is hard to obtain through animal immunization (hard-to-drug epitopes).
Challenges¶
The search space for such a library design is huge as we need to determine the amino acid types and positions of more than ten residues in the paratope while considering the possible intra-antibody and epitope-paratope interactions. Traditional methods could hardly balance efficiency and accuracy. Generative AI and geometric deep learning technologies, however, have achieved new state-of-the-art in the success rates of antibody design.
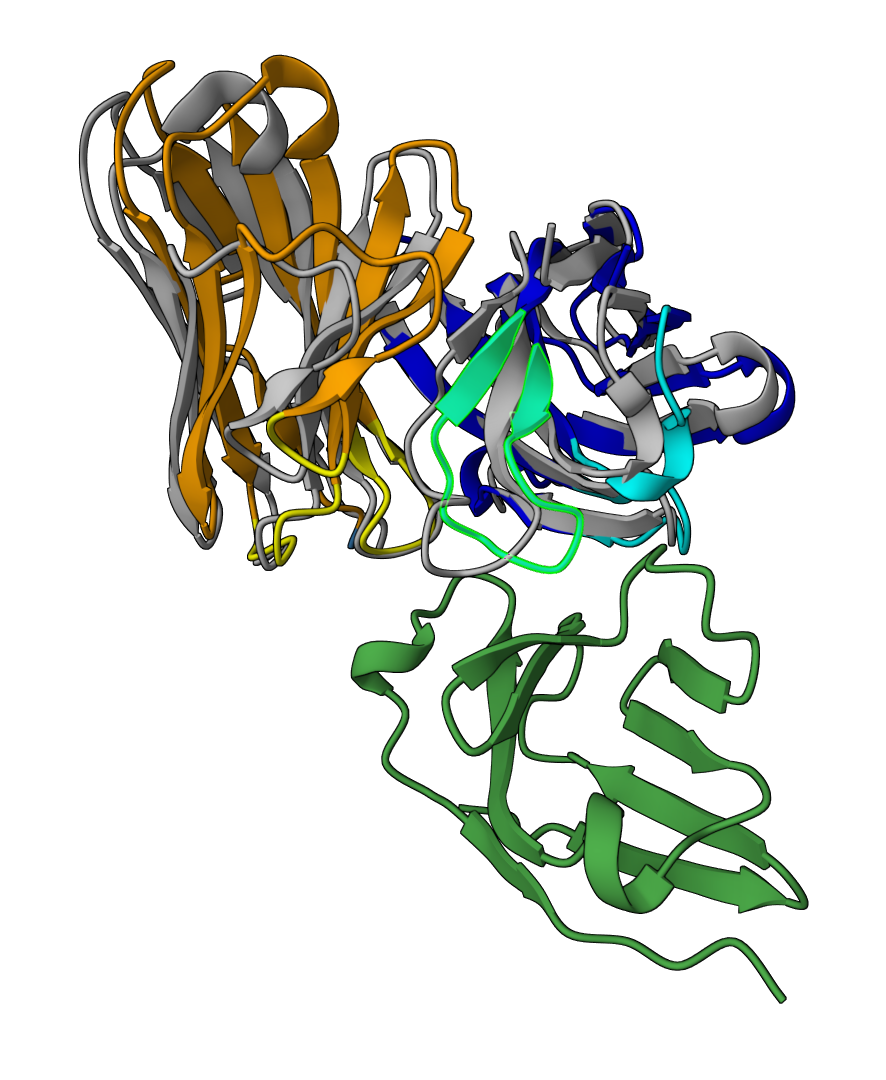
How this job works¶
Here we provide a high-level overview of how this job works to help you understand the parameters and results better and achieve better design outcomes.
- De novo design: Based on our generative model, sequences and structures of the specified CDR residues are designed de novo according to the target epitope to maximize the antibody-antigen interaction.
- More sequence design: To further increase the sequence diversity, we generate more sequences from the previously designed Ag:Ab structures.
- Virtual screening: A virtual screening module is built into our design pipeline to identify the most promising candidates. The screening criteria can be customized by you in the result page.
Inputs¶
To submit a Targeted CDR Library Design job, open the Project Editor and click "New Job" button on the left sidebar. Then click "Targeted CDR Library Design" under the "Antibody Design" group to open the job submission page.
-
Antigen: The target antigen.
- Structure: Choose any PDB or mmCIF file containing the desired antigen structure. If the file is already open in the structure viewer, click "Select from viewer". If the file is in your current project, click "Select from project". Otherwise, you can upload it in the Files & Jobs or import it from cloud databases like PDB.
- Chain: Specify the chain ID(s) of the antigen, e.g., "A,B".
Multi-chain antigen
For the current version, we recommended you to select a single chain for the antigen, as the current model might change the relative positions of the multiple antigen chains. We plan to fix this problem in future versions.
- Epitope: A set of residues comprising the epitope (antibody-binding sites) of the antigen. Input a comma-separated list of contiguous sequence fragments with the format
{chain}:{start_res}-{end_res}, e.g.,A:2-10,A:15-30,B:40-100. See Epitope Format for more details. You could also select the epitope from the antigen structure using the Structure Viewer and click on "import from selection" to fill in this input box automatically.
Import epitope from selection
Click the "Epitope" input box and you will see "open file" or "import from selection" below the input box.
- If the input structure file is not opened in the Structure Viewer, you can click "open file" to open it.
- If the input structure file is already open in the Structure Viewer, you can enter Selection Mode and select mutation sites on the structure. Then click "import from selection" to automatically fill this input box.
- You can also run the Interface Visualization job, which will create components in the Structure Viewer. You can then select a component and click "import from selection" to automatically fill this input box.
Epitope selection and structural diversity
First, please note that the expected input is not the binding sites of the designed CDR residues, but rather the binding sites of the entire antibody.
A smaller epitope selection will tend to yield less structural diversity. If you want to design a library with more structural diversity, try to provide an epitope with at least 6 residues. However, an epitope selection too large may result in a low success rate, since the model will try to design antibodies that can bind to most of the selected epitope residues. A nice work around is launching multiple design jobs with different epitope selections.
-
Antibody: An antibody with the CDR region to be designed.
- Antibody Name: Name of the antibody. Defaults to "Antibody". To change it, hover above the antibody name, and click on the "
 " button.
" button. - Format: Choose the format of the antibody from "VH+HL" (normal antibodies, the default) and "VH" (single-domain antibodies or nanobodies).
- Sequence: Sequence of the antibody template. The regions to be designed should be replaced by the placeholder
[{region}, {init_seq}]. Below is an example.
Specifying the region to be designed
{region}tag specifies the region to be designed (H1/H2/H3/L1/L2/L3)- H1/2/3 tags can only appear in the heavy chain, and L1/2/3 only in the light chain.
- Each region tag can appear only once, i.e. there can not be two "[H3, ...]" tags in the input.
- Currently, only regions in one chain can be designed at the same time. That means you can design H1, H2 and H3 at the same time, but can not design H3 and L3 at the same time. Note that the success rate of design may decrease when there are more regions/residues to design.
{init_seq}is the initial sequence of the region, with X denoting residues to be designed.- For example,
[H1, XXGSX]specifies 3 residues to be designed in the H1 region. The residues "GS" are fixed during design. - If all residues in the region are to be designed, simply input the length of the region as
{init_seq}, e.g.[H3, 11]specifies 11 residues to be designed in the H3 region. - Currently, each job can only design antibodies of the same length. If you want to design antibodies of varying lengths, please launch multiple jobs.
 (Upload Sequence): Upload your own antibody template (a .FASTA file) by clicking the "" button. If the format is "VH+VL", your antibody template must have 2 chains with labels ending with "_H" and "_L" respectively. Above is an example. If the format is "VH", your antibody template must have only one chain with a label ending with "_H".
(Upload Sequence): Upload your own antibody template (a .FASTA file) by clicking the "" button. If the format is "VH+VL", your antibody template must have 2 chains with labels ending with "_H" and "_L" respectively. Above is an example. If the format is "VH", your antibody template must have only one chain with a label ending with "_H".
- Antibody Name: Name of the antibody. Defaults to "Antibody". To change it, hover above the antibody name, and click on the "
-
Job Name: Name of the job. Note that the job name must be unique within the project.

Epitope Format¶
In GeoBiologics, the epitope residue list comprises one or more "sites" and "fragments".
- Each site is represented as
{chain_id}:{res_id}, e.g.,H:100. - Each fragment is represented as
{chain_id}:{start_res_id}-{end_res_id}. For example,H:100-112. Note that the start and end residue IDs must not include insertion codes to avoid ambiguity. For example, certain antibodies may have the following IMGT numbering: "110 111 111A 112B 112A 112", in this case, if you specifyH:110-112, we will be unable to determine which residues you intend to mutate, so it is recommended to use site representation in this case.
Multiple sites or fragments can be joined by commas, e.g., H:100-110,H:111,H:111A,H:112B,L:50-60,L:92.
Models & Parameters¶
GeoFlow-Design, our proprietary generative model for antibody design, is available for this job. Parameters of this model are as follows.
- # designs: The number of structures to generate (default to 100). Note that the number of sequences generated is about three times this number (as shown in the Task Summary section), and the number of sequences passing the virtual screening filter is usually less than this number. In real-world projects, you are recommended to try a ~200 design run first to validate the parameters, and then adjust the number of designs according to the success rates of the virtual screening in the first run.
- Temperature: Temperature for antibody sequence sampling (default to 1). Lower temperature leads to more diverse structural designs. Recommended to be between 0.8 and 1.2.
- seed: Random seed for structure initialization (default to a random integer).
Results¶
Click the job name in the Files & Jobs panel to view the job results.
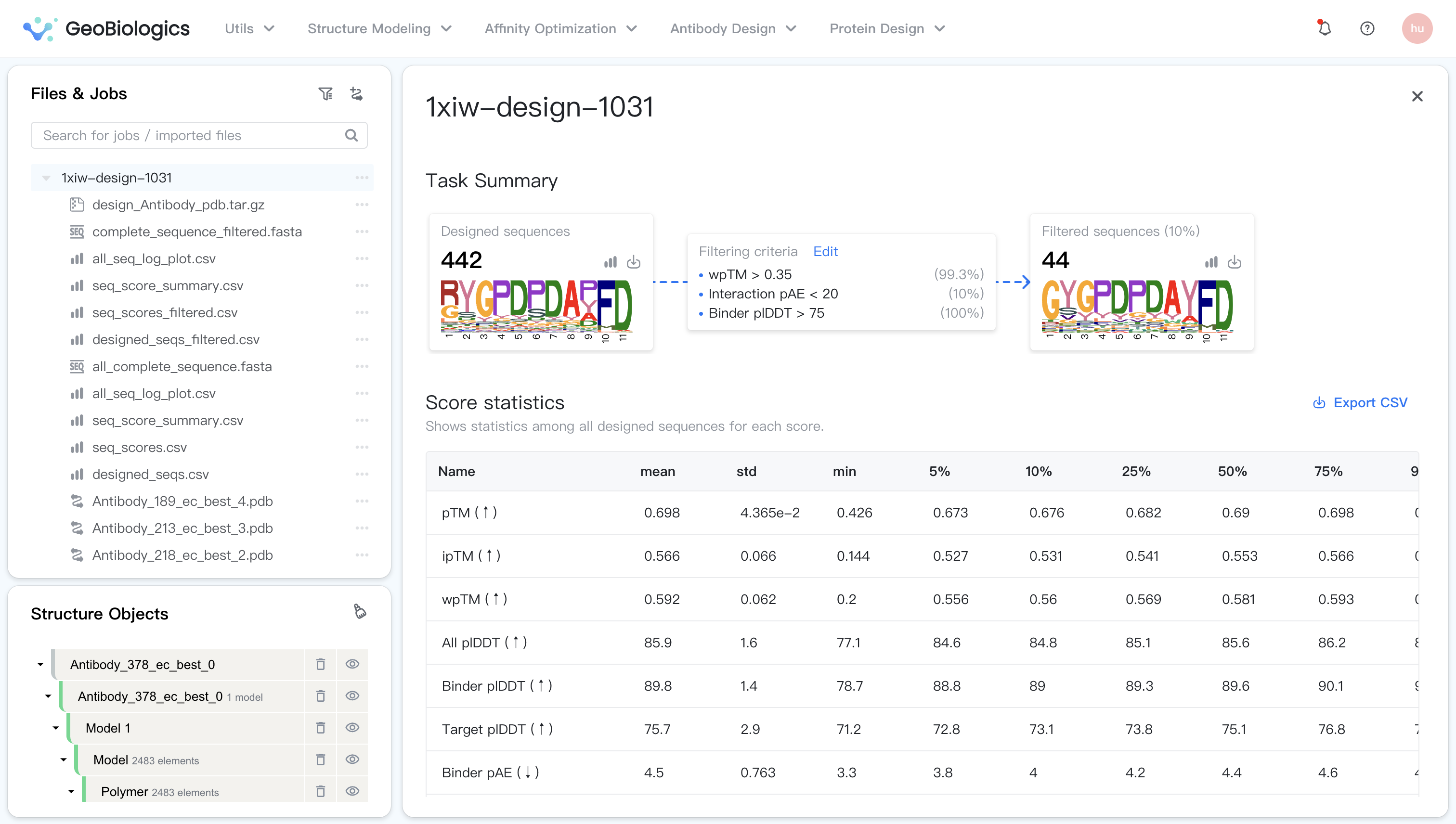
Task Summary¶
The task summary displays key information about the designed sequences, the filtering criteria, and the filtered sequences. Let's break it down here:
- Designed sequences: The bold number shows the total number of designed sequences.
 : Click to preview the first 500 designed sequences and their scores.
: Click to preview the first 500 designed sequences and their scores. : Hover to download (1) sequences of the designed region(s) and all corresponding scores in a .csv file; (2) the full designed sequences in a .FASTA file; and (3) all the designed sequences in a .tar.gz file.
: Hover to download (1) sequences of the designed region(s) and all corresponding scores in a .csv file; (2) the full designed sequences in a .FASTA file; and (3) all the designed sequences in a .tar.gz file.- Logo plot: A convenient logo plot visualizing the amino acid distribution of the designed sequences.
- Filtered sequences: The bold number shows the total number of filtered sequences. The percentage is the ratio of designed sequences passing the virtual screening filter.
- : Click to preview the first 500 filtered sequences and their scores.
- : Click to download (1) filtered sequences of the designed region(s) and all corresponding scores in a .csv file; (2) the full filtered sequences in a .FASTA file.
- Filtering criteria: The filtering criteria used in the virtual screening. Each criterion is accompanied by a percentage to the right, indicating the ratio of sequences passing the criterion. You can customize the filtering criteria by clicking on "Edit". The filtered sequences will be updated accordingly.
Scores computed during virtual screening
The virtual screening process is based on our proprietary GeoFlow model, which checks the consistency between the designed sequence and structures by calculating the following scores:
- pTM: Predicted TM (Template modeling) score for the generated Ab:Ag complex. Higher score indicates higher overall confidence in the predicted complex structure. Note that this score is an average of the entire complex so it can be easily dominated by, e.g., a large domain.
- ipTM: The predicted interface TM-score for the Ab:Ag complex. Higher score indicates higher confidence in the interface regions.
- wpTM: The predicted weighted TM-score for the Ab:Ag complex, calculated by \(\mathrm{wpTM} = 0.8 \times \mathrm{ipTM} + 0.2 \times \mathrm{pTM}\). Generally a reliable metric to rank/screen antibody sequences. Higher score indicates higher overall confidence in the Ab:Ag complex.
- All plDDT: Predicted lDDT (local Distance Difference Test) score averaged over the entire Ab:Ag complex. Higher score indicates better overall structure consistency.
- Binder plDDT: The predicted lDDT score averaged over the antibody. Higher score indicates better antibody structure consistency.
- Target plDDT: The predicted lDDT score averaged over the antigen. Higher score indicates better antigen structure consistency.
- Binder pAE: The predicted aligned error (AE) averaged over the antibody-antibody residue pairs. Lower score indicates better antibody structure consistency.
- Target pAE: The predicted aligned error (AE) averaged over the antigen-antibody residue pairs. Lower score indicates better antigen structure consistency.
- Interaction pAE: The predicted aligned error (AE) averaged over the antibody-antigen residue pairs. Lower score indicates higher confidence in the predicted antibody-antigen interaction.
- Binder-aligned lRMSD: The antibody's root-mean-square deviation (RMSD) when aligning the predicted antibody structure to the designed structure. Lower score indicates better antibody structure consistency. This score is usually low.
- Target-aligned lRMSD: The antibody's root-mean-square deviation (RMSD) after aligning the targets (antigens) in the predicted complex structure to those in the designed structure. Lower score indicates better epitope consistency. This score can be high if the predicted epitope (antibody-binding sites) is far from the designed structure.
Score statistics¶
This section shows the score statistics of all designed sequences. The statistics shown include the mean, standard deviation, minimum, and maximum scores, and the 5%, 10%, 25%, 50%, 75%, 90% and 95% percentiles. You may use these statistics to understand the distribution of the scores and to set the filtering criteria.
Representative structures¶
To help you visualize the designed results, we cluster all designed structures according to the epitope and show you the representative structures for each cluster. Note that the epitope residues are numbered as in the output structure, which might differ from the numbering in the input structure. You may click "open" to open the structures in the Structure Viewer.