Protein Inverse Folding¶
Sequence design is the task of generating a sequence that is compatible with a given protein/antibody structure. It is of great importance in medicine (for example, in drug design) and biotechnology (for example, in the design of novel enzymes).
Features¶
-
Validated algorithm: The ProteinMPNN algorithm is experimentally validated and widely used for protein sequence design. It can also be used to design antibodies after some fine-tuning.
-
Cloud-based, Mass-scale: The algorithm is deployed on our cloud platform and can be run on a massive scale. Results are stored in a CSV file and can be easily downloaded for further analysis.
Inputs¶
To submit a Protein Inverse Folding job, open the Project Editor and click "New Job" button on the left sidebar. Then click "Protein Inverse Folding" under the "Protein Design" group to open the job submission page.
- Target: The target structure for sequence design.
- Structure: Target structure in PDB/mmCIF format. The structure can be uploaded from a local machine, imported from a cloud database or generated by a GeoBiologics job.
- Chains to design: IDs of the chains to design. If not specified, all chains will be designed.
- Job Name: Name of the job. Note that the job name must be unique within the project.

Models & Parameters¶
Two models are available for this job: ProteinMPNN, suitable for general protein design, and ProteinMPNN-Ab, more adept at antibody design.
The parameters for the two models are the same, as shown below.
- # designs: The number of sequences to generate. The default value is 10.
Results¶
Click the job name in the Files & Jobs panel to view the job results.
The results are stored in a CSV file, which can be downloaded by clicking the "![]() " button to the top-right of the Summary Table.
" button to the top-right of the Summary Table.
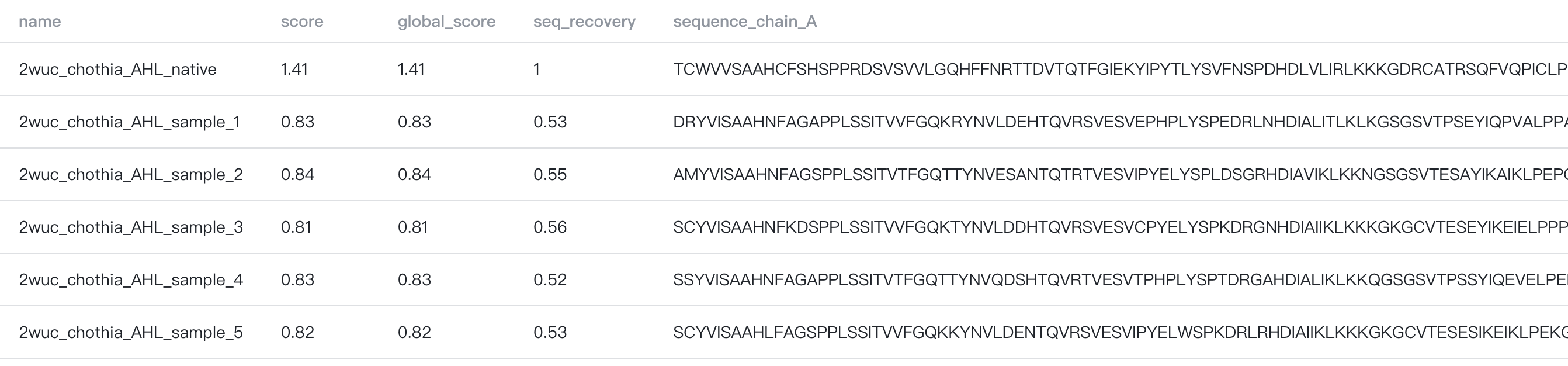
The summary table contains the following columns:
- name: name of the output sequence
- score: average negative log likelihood of the sampled (designed) amino acids
- global_score: average negative log likelihood of amino acids in all chains
- seq_recovery: average sequence identity between the designed sequence and the input sequence
- sequence_chain_{chain_id}: designed sequence for chain {chain_id}. E.g., if you chose
H,Las the chains to design, you would have two columns in the summary table,sequence_chain_Handsequence_chain_L.