Protein Optimization¶
Protein optimization is the process of optimizing the properties of a protein, including Fitness, Thermostability and Solubility. By analyzing the impact of single or multiple point mutations of proteins on their properties, users can form targeted evolution strategies to accelerate protein engineering campaigns.
Inputs¶
To submit an Protein Optimization job, please open the Project Editor and click "New Job" button on the left sidebar. Then click "Protein Optimization" under the "Protein Design" group to open the job submission page.

-
Input Mode: You can select one of the following input modes:
- Sequence-based: Structure will be predicted from the protein sequence with ESMFold. Only select this option if the protein structure is not available.
- Protein Sequence: The protein sequence to be optimized, with a maximum length of 1000 amino acids.
- Structure-based: Use protein sequence and structure to provide more reliable results.
- Structure selector: Select the structure to optimize from the project or from the current viewer.
- Chain to opt.: The chain ID in the structure to optimize.
- Sequence-based: Structure will be predicted from the protein sequence with ESMFold. Only select this option if the protein structure is not available.
-
Mut. mode: The mutation mode can be either "Saturation Mutagenesis" or "Custom". "Saturation Mutagenesis" indicates single-site saturation mutations, while "Custom" allows for custom single or multiple combinations of mutations.
-
Mutation sites: A comma-separated list of mutation sites (1-based), e.g.
32-35,100,102-105, where32-35represents a closed interval, including sites 32, 33, 34, and 35. -
Mutation list: A mutation list where each line represents a mutant carrying one or more mutations, e.g.,
G25T(single mutant) orA35W,Q64F,E103T(triple mutant). Multiple mutations are separated by commas.
-
-
Job Name: The name of the job. Please note that the job name must be unique within the project.
Models & Parameters¶
You can use our proprietary GeoProt model to run this job. The parameters of the model are as follows:
-
Objective: You can select one or more optimization objectives from "Fitness" "Thermostability" and "Solubility"
- "Fitness" is a general metric for protein survival and function;
- "Thermostability" is the protein's stability under heated conditions.
- "Solubility" is the protein's solubility in water.
Results¶
Click Job Results in the Files & Jobs panel to view the job results.
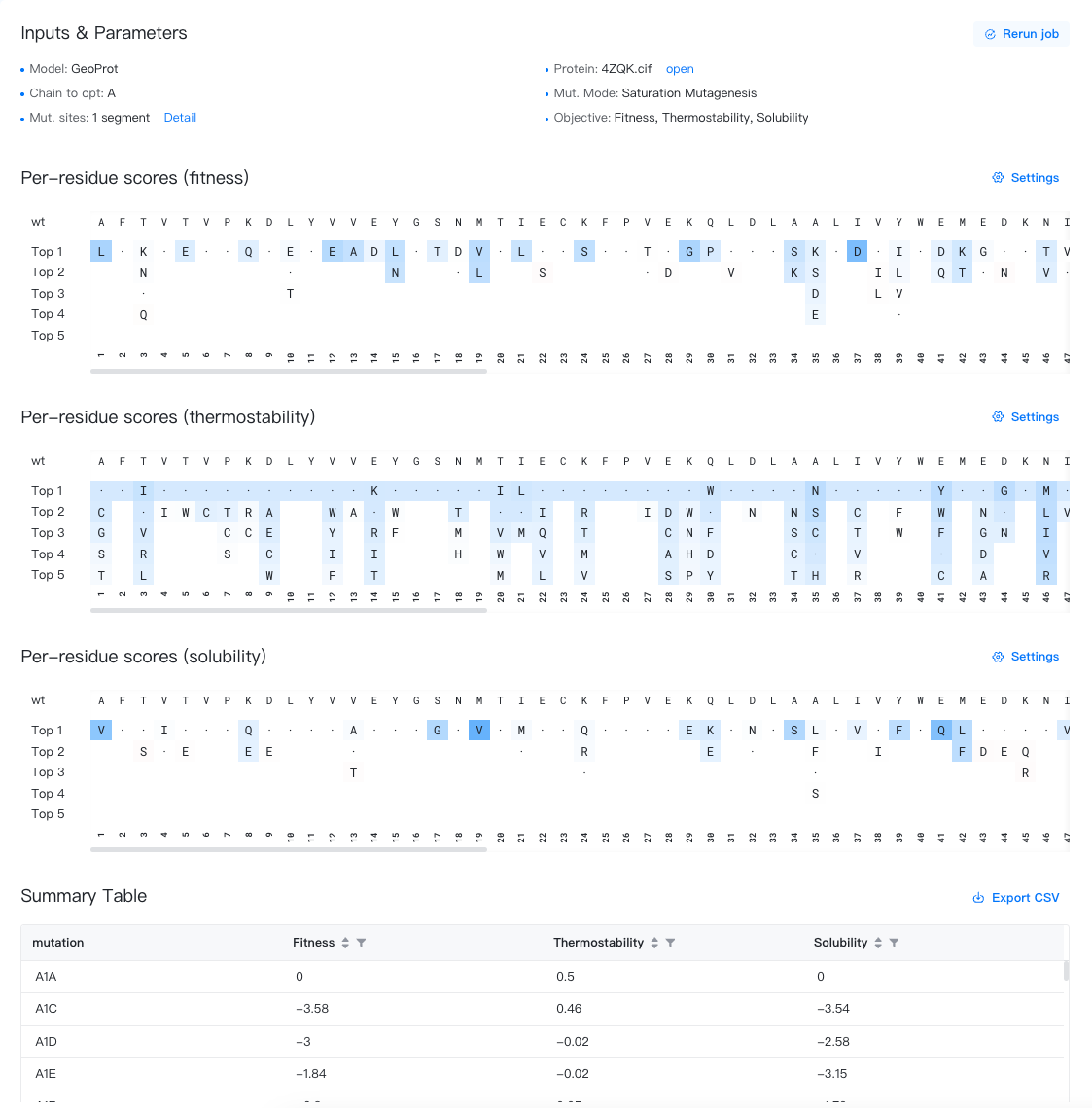
Summary Table¶
The results are stored in a CSV file, which can be downloaded by clicking the "![]() " button to the top-right of the Summary Table.
" button to the top-right of the Summary Table.
The summary table contains the following columns:
-
Mutation: Labels of the point mutations, formatted as "original amino acid + residue ID + mutated amino acid", e.g., F103Y. The first row is fixed as "WT", representing the wild-type enzyme before mutation.
-
Fitness: Changes in the fitness due to mutations; a score greater than 0 indicates improved fitness; a score less than 0 indicates decreased fitness.
-
Thermostability: Changes in the thermostability due to mutations; a score greater than 0 indicates improved thermostability; a score less than 0 indicates decreased thermostability.
-
Solubility: Changes in the solubility due to mutations; a score greater than 0 indicates improved solubility; a score less than 0 indicates decreased solubility.
Mutation Suggestions¶
If the mutation mode selected is "Saturation Mutagenesis", the results page will also include a section "Per-residue scores", showing the top 5 amino acids recommended for each mutation site.
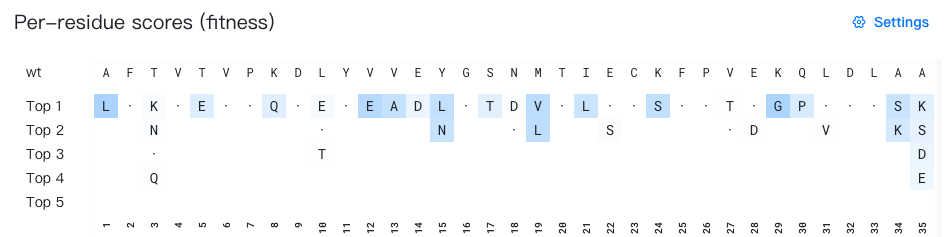
- The "wt" row shows the wild-type amino acid for each mutation site.
-
"Top 1-5" lists the top 5 recommended amino acids for each site.
Each cell displays a mutant amino acid type. Self-mutations are marked with "·" for easier identification. Mutations with low probability is hidden by default.
The background color of each cell shows the advantage of the current mutation.
- In the "raw score" mode (the default mode), the fitness-enhancing mutations are colored in blue, and the fitness-weakening mutations are colored in red.
- In the "per-site probability" mode, the mutations with high per-site probability are colored in purple. Note that in this mode, probabilities across sites are not comparable.
When you hover over a cell, a tooltip (e.g. "3 | T → K | score = 0.17") will appear showing the wild-type and mutant amino acids, as well as the predicted score value for the current mutation.
For a detailed documentation of this section, please refer to Mutation Advisor.