Protein Sequence Design¶
Sequence design is the task of generating a sequence that is compatible with a given protein/antibody structure.
In this task, we first generate sequences based on the target structures, and then use AF2-Multimer to predict the structure of the designed sequences. The self-consistent sequences (i.e. the sequences that are likely to fold into the target structures) are then selected for wet-lab validation.
Features¶
- Validated algorithm: The ProteinMPNN algorithm is published in Science, validated in numerous in silico protein design campaigns and widely used for protein sequence design.
- Customized design: Set your amino acid preferences and solubility preferences to design sequences that are more likely to meet your needs.
- Cloud-based, Mass-scale: The algorithm is deployed on our cloud platform and can be run on a massive scale. Results are stored in a CSV file and can be easily downloaded for further analysis.
Inputs¶
To submit a Protein Sequence Design job, open the Project Editor and click "New Job" button on the left sidebar. Then click "Protein Sequence Design" under the "Protein Design" group to open the job submission page.
- Job Name: Name of the job. Note that the job name must be unique within the project.
- Designed sequence: Here we specify the segments within the target structure to design.
- Target Structures: The structures we want the sequence to fold into, in PDB/mmCIF format. The structures can be uploaded from a local machine, imported from a cloud database (see Add File to Project for instructions) or generated by a GeoBiologics job.
- Target Contigs: This is a non-editable field generated from the target structures. It contains the contigs description of the target structures (they must have the same contigs, otherwise an error will be raised).
- Design Contigs: Here we specify the segments within the target structure to design as designed contigs, and the segments to fix as fixed contigs.
- Fixed contig: The sequence of this segment will be copied from the target structure. Represented as
{chain_id}:{start_resi_id}-{end_resi_id}or simply{chain_id}:{resi_id}. - Designed contig: The sequence of this segment will be designed de novo. Represented as
{min_len}-{max_len}or simply{len}.
- Fixed contig: The sequence of this segment will be copied from the target structure. Represented as
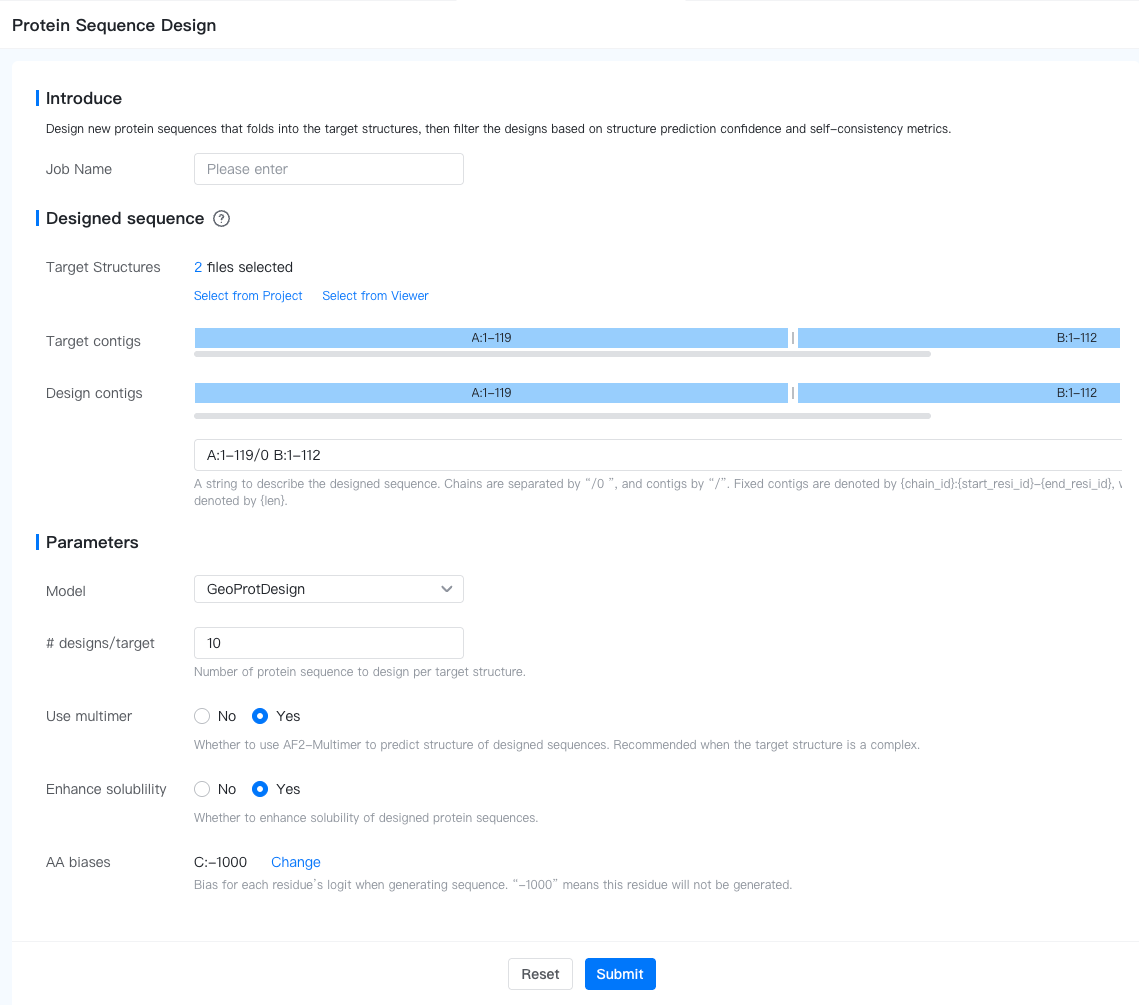
Models & Parameters¶
GeoProtDesign is a sequence design model based on ProteinMPNN and validated in multiple protein design campaigns.
The parameters for the model are as shown below.
- # designs/target: The number of sequences to generate for each target structure. The default value is 10.
- Use multimer: Whether to use AF2-Multimer to predict structure of designed sequences. Recommended when the target structure is a complex.
- Enhance solubility: Whether to enhance solubility of designed protein sequences.
- AA biases: Bias for each residue’s logit when generating sequence. “-1000” means this residue will not be generated.
Results¶
Click the job name in the Files & Jobs panel to view the job results.
Task Summary¶
The task summary displays key information about the designed sequences, the filtering criteria, and the filtered sequences. Let's break it down here:
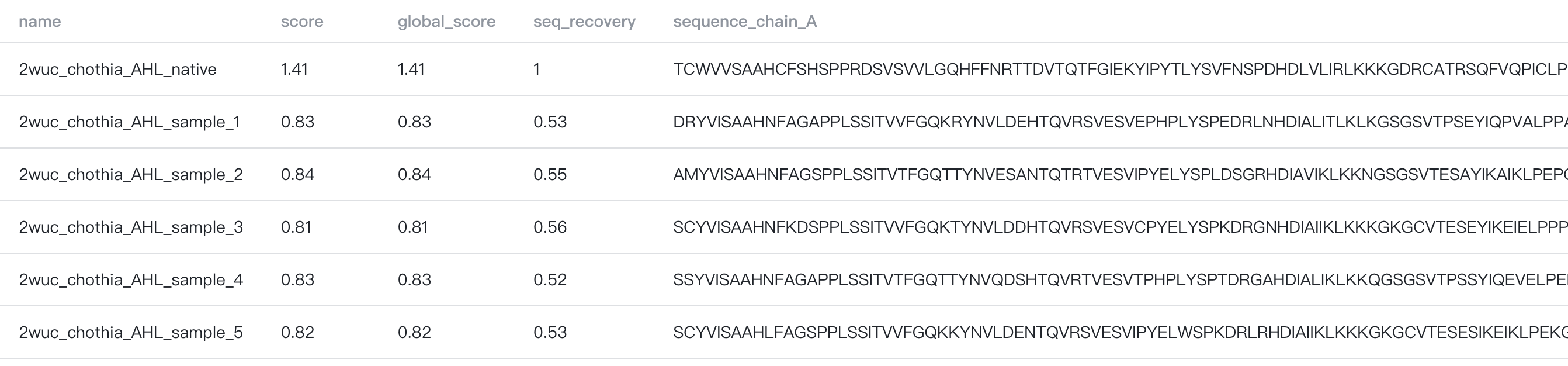
- Designed sequences: The bold number shows the total number of designed sequences.
 : Click to preview the first 500 designed sequences and their scores.
: Click to preview the first 500 designed sequences and their scores. : Hover to download (1) sequences of the designed region(s) and all corresponding scores in a .csv file; (2) the full designed sequences in a .FASTA file; and (3) all the designed sequences in a .tar.gz file.
: Hover to download (1) sequences of the designed region(s) and all corresponding scores in a .csv file; (2) the full designed sequences in a .FASTA file; and (3) all the designed sequences in a .tar.gz file.- Logo plot: A convenient logo plot visualizing the amino acid distribution of the designed sequences.
- Filtered sequences: The bold number shows the total number of filtered sequences. The percentage is the ratio of designed sequences passing the virtual screening filter.
- : Click to preview the first 500 filtered sequences and their scores.
- : Click to download (1) filtered sequences of the designed region(s) and all corresponding scores in a .csv file; (2) the full filtered sequences in a .FASTA file.
- Filtering criteria: The filtering criteria used in the virtual screening. Each criterion is accompanied by a percentage to the right, indicating the ratio of sequences passing the criterion. You can customize the filtering criteria by clicking on "Edit". The filtered sequences will be updated accordingly.
Scores computed during virtual screening
The virtual screening process checks the consistency between the designed sequence and structures by calculating the following scores:
- mpnn: The sequence design negative log-likelihood. Smaller score indicates higher sequence likelihood.
- plDDT: Predicted lDDT (local Distance Difference Test) score averaged over the entire Ab:Ag complex. Higher score indicates better overall structure consistency.
- pTM: Predicted TM (Template modeling) score for the generated Ab:Ag complex. Higher score indicates higher overall confidence in the predicted complex structure. Note that this score is an average of the entire complex so it can be easily dominated by, e.g., a large domain.
- ipTM: The predicted interface TM-score for the complex. Higher score indicates higher confidence in the interface regions. For monomer proteins, this is always 0.
- pAE: The predicted aligned error (AE) averaged over all residue pairs. Lower score indicates higher confidence in the predicted structure.
- RMSD: The root-mean-square deviation (RMSD) when aligning the predicted structure to the designed structure. Lower score indicates better sequence-structure consistency.
Score statistics¶
This section shows the score statistics of all designed sequences. The statistics shown include the mean, standard deviation, minimum, and maximum scores, and the 5%, 10%, 25%, 50%, 75%, 90% and 95% percentiles. You may use these statistics to understand the distribution of the scores and to set the filtering criteria.