Antibody-Antigen Docking¶
Antibody-antigen docking predicts the structure of an antibody-antigen complex based on their sequences, helping users understand how the antibody and antigen bind and the location of binding sites.
In practice, due to prediction errors, the multiple predictions are returned. Each model are accompanied by quality control metrics to help you grade the reliability of the prediction.
Challenges & Features¶
The large size of protein systems makes determining the optimal orientation of two proteins challenging. Additionally, flexible regions often undergo conformational changes during docking. GeoBiologics' Antibody-Antigen Docking module has the following advantages:
-
State-of-the-art Algorithm: Utilizing the latest geometric deep learning techniques and large-scale structural pretraining, GeoFlow V2 demonstrates strong and robust performance in antibody-antigen docking, with a success rate ~30% higher than AlphaFold3 reproduction models and ~93% higher than AlphaFold2.
-
Versatile Constraint Support: GeoFlow V2 enables you to input the epitope on the antigen and partial antigen structure to guide the docking results towards better accuracy. The docking success rate can increase from ~45% to ~75% given 4 accurate epitope residues.
-
Easy Analysis: We know your pain of browsing through tons of predicted structures. The predicted structures are automatically aligned by the antigen and clustered by epitope regoins. All structures come with a series of quality control metrics to help you grade the reliability of the prediction. When you open a predicted structure, the Interface Visualization panel will automatically pop out, listing all antibody-antigen interactions for your analysis.
Inputs¶
To submit an Antibody-Antigen Docking job, open the Project Editor and click "New Job" button on the left sidebar. Then click "Antibody-Antigen Docking" under the "Structure Modeling" group to open the job submission page.
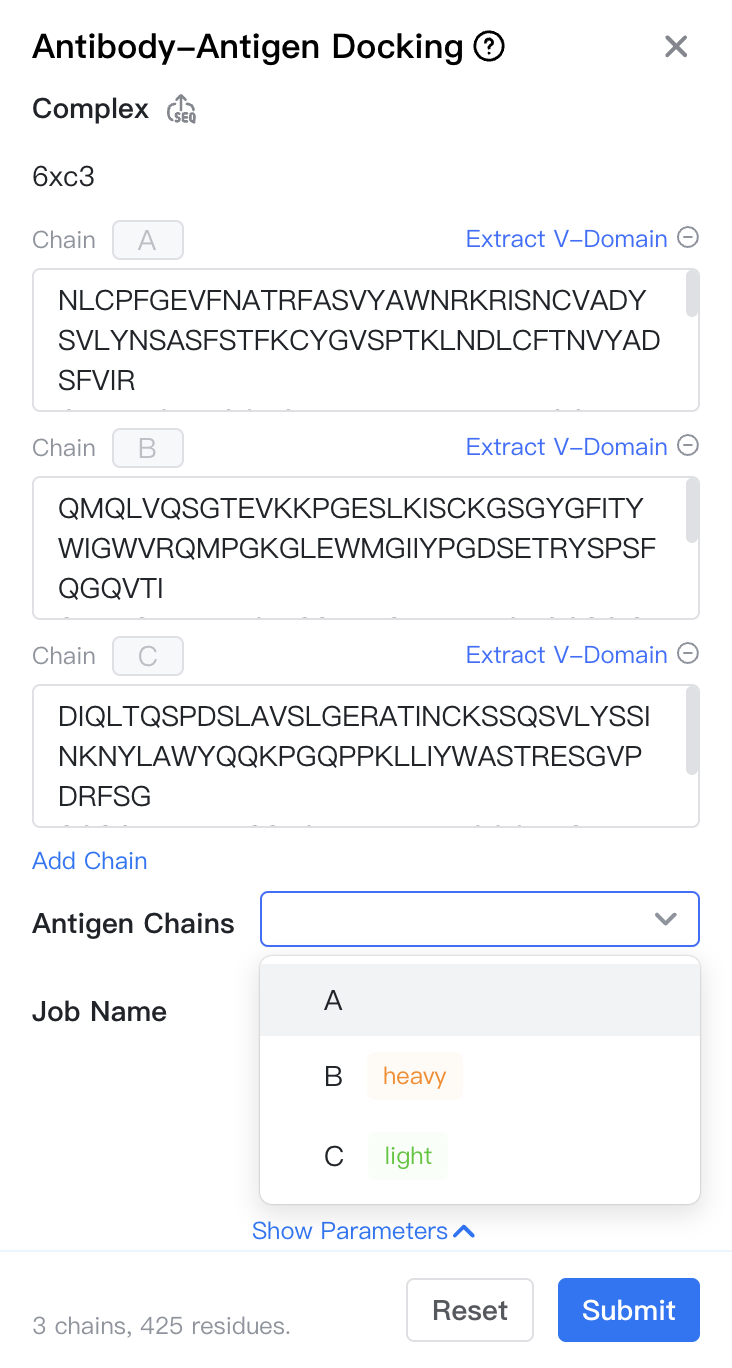
-
Complex: The sequence of the antibody-antigen complex.
-
Upload a FASTA file: Click the "
 " button to upload your own antibody-antigen complex (a .FASTA file). The file can only contain one antibody-antigen complex, and the FASTA labels of the chains must have the same prefix and end with
" button to upload your own antibody-antigen complex (a .FASTA file). The file can only contain one antibody-antigen complex, and the FASTA labels of the chains must have the same prefix and end with _{chain_id}. -
Copy FASTA strings: Copy the FASTA string content directly into the sequence input box. The platform will automatically parse and create a multi-chain sequence. The FASTA string must meet the above requirements. You can input the following sequence to test this feature:
>6xc3_C NLCPFGEVFNATRFASVYAWNRKRISNCVADYSVLYNSASFSTFKCYGVSPTKLNDLCFTNVYADSFVIR GDEVRQIAPGQTGKIADYNYKLPDDFTGCVIAWNSNNLDSKVNYNYLYRLFRKSNLKPFERDISTEIYQA GSTPCNGVEGFNCYFPLQSYGFQPTNGVGYQPYRVVVLSFELLHAPATVCGPK >6xc3_H QMQLVQSGTEVKKPGESLKISCKGSGYGFITYWIGWVRQMPGKGLEWMGIIYPGDSETRYSPSFQGQVTI SADKSINTAYLQWSSLKASDTAIYYCAGGSGISTPMDVWGQGTTVTVSS >6xc3_L DIQLTQSPDSLAVSLGERATINCKSSQSVLYSSINKNYLAWYQQKPGQPPKLLIYWASTRESGVPDRFSG SGSGTDFTLTISSLQAEDVAVYYCQQYYSTPYTFGQGTKVEIK- Direct Input: Input the sequence of each chain in the sequence input box. Note that the chain IDs are fixed as ABCDEF... sequentially.
-
-
Antigen Chains: Select which chains belong to the antigen; the remaining chains will default to the antibody.
Chain Type Recognition
GeoBiologics will automatically recognize and label the V-domain structure in the structure, including the antibody heavy chain (heavy), antibody light chain (light), TCR alpha chain (alpha), and TCR beta chain (beta).
-
Provide Structure: Click "provide structure" below the chain ID to open a panel to provide full/partial structure of the antigen.
- Reference Structure: Select the reference structure of the antigen.
- Input Sequence: The sequence of the antigen. This is synchronized with the sequence input box.
- Structrue/Sequence Segment: Match the label_seq_id in the reference structure to the sequence IDs in the sequence. Label_seq_id can be obtained by (1) selecting the segment in the reference structure, (2) hovering on the
 button and (3) clicking "Copy selected sites (label)".
button and (3) clicking "Copy selected sites (label)".

Job submission form: Provide Structure -
Constraints: Specify antigen residues (identified by their chain IDs and sequence IDs) that should bind to a specific antibody chain in the "Constraints" section.
-
Job Name: The name of the job. Note that the job name must be unique within the project.
Models & Parameters¶
You can use our proprietary GeoFlow V2 model for this task. The parameters of this model are as follows:
- Mode: You can choose between "fast" and "accurate". The former is faster, while the latter samples more docking poses and has a higher docking success rate.
Results¶
Click Job Results in the Files & Jobs panel to view the job results.
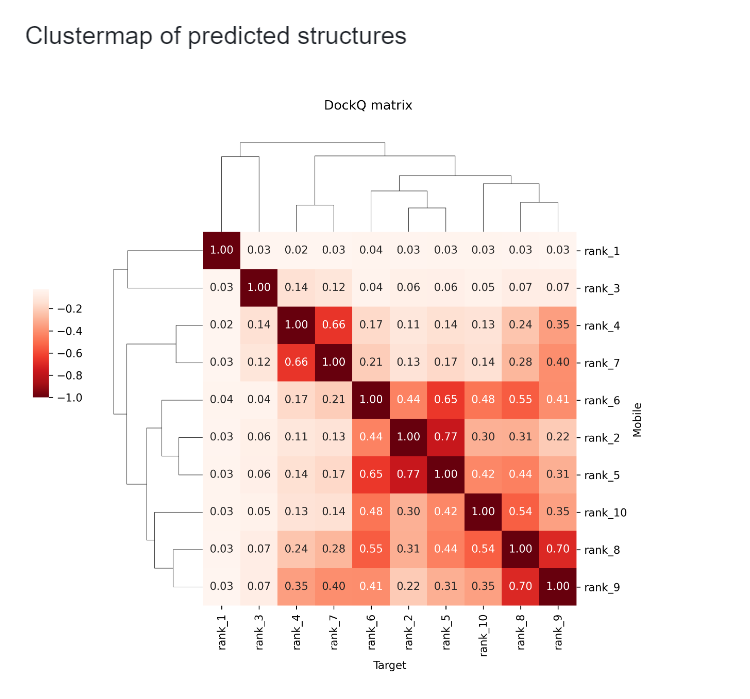
Clustermap of Predicted Structures¶
This section displays the clustering results of predicted structures. Each square in the clustermap is annotated with the ligRMSD value between two predicted structures, where ligRMSD is the RMSD value of two antibody structures when their corresponding antigens are aligned. Through this clustermap, you can easily identify which structures are similar and which are different, without the need to manually check each structure. You can also use the "cluster_label" column to view the cluster ID each structure belongs to.
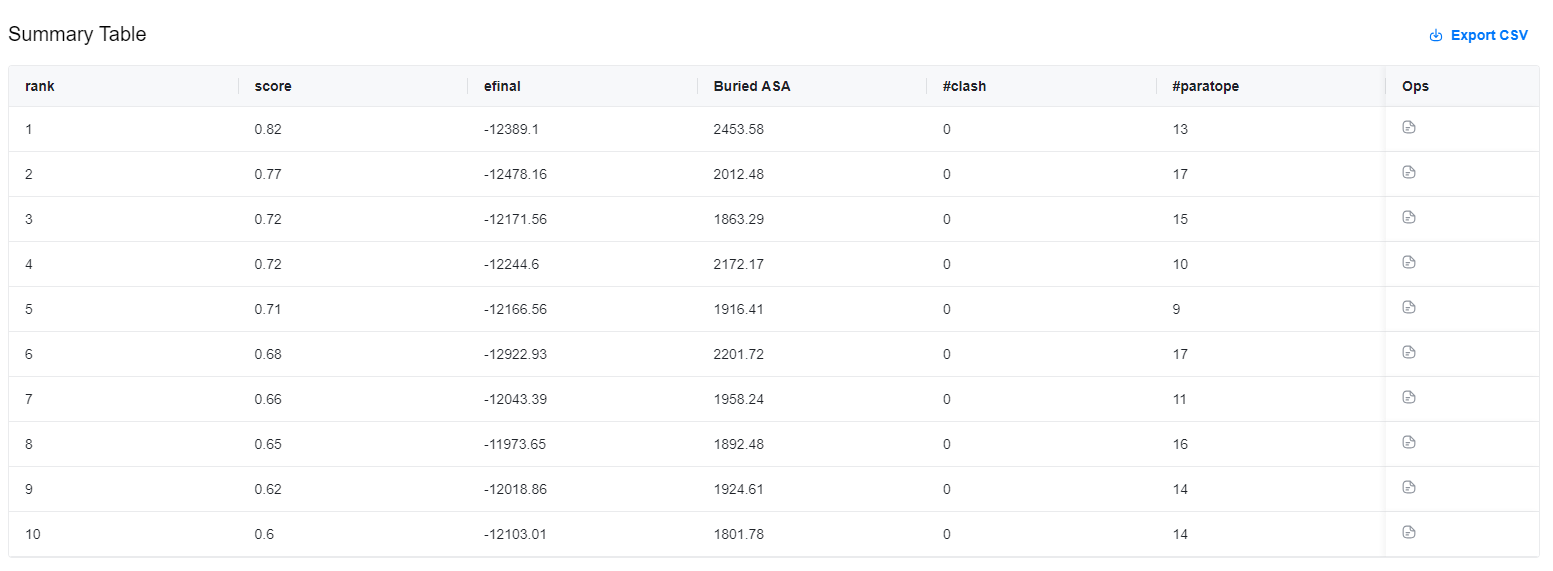
Summary Table¶
The summary table shows the confidence and various metrics of the 10 predicted structures, including:
- name: Name of the predicted structures, containing ranks starting from 0.
- ranking_score: Model's confidence in this prediction. Generally, if score > 0.8, the structure is considered reliable; if score < 0.5, it is considered unreliable.
- E_init: Energy before minimization predicted by the AMBER99 model, with lower values being better.
- E_final: Energy after minimization predicted by the AMBER99 model, with lower values being better.
- Buried ASA: Reduction in solvent-accessible surface area due to antigen-antibody binding. For reliable structures, this value is usually > 1200.
- antigen_contacts: Number of antigen residues that are in contact with the antibody.
- cdr_contacts: Number of CDR residues that are in contact with the antigen.
- cluster_label: Cluster label of the predicted structure. Within each cluster, the ligRMSD value is usually < 8 Å.
- ipTM: Predicted TM-score for the antibody-antigen interface. Higher is better.
- ipAE: Predicted aligned error for the antibody-antigen interface. Lower is better.
Results are stored in a CSV file, which can be downloaded by clicking the "![]() " button in the top-right corner of the results table.
" button in the top-right corner of the results table.
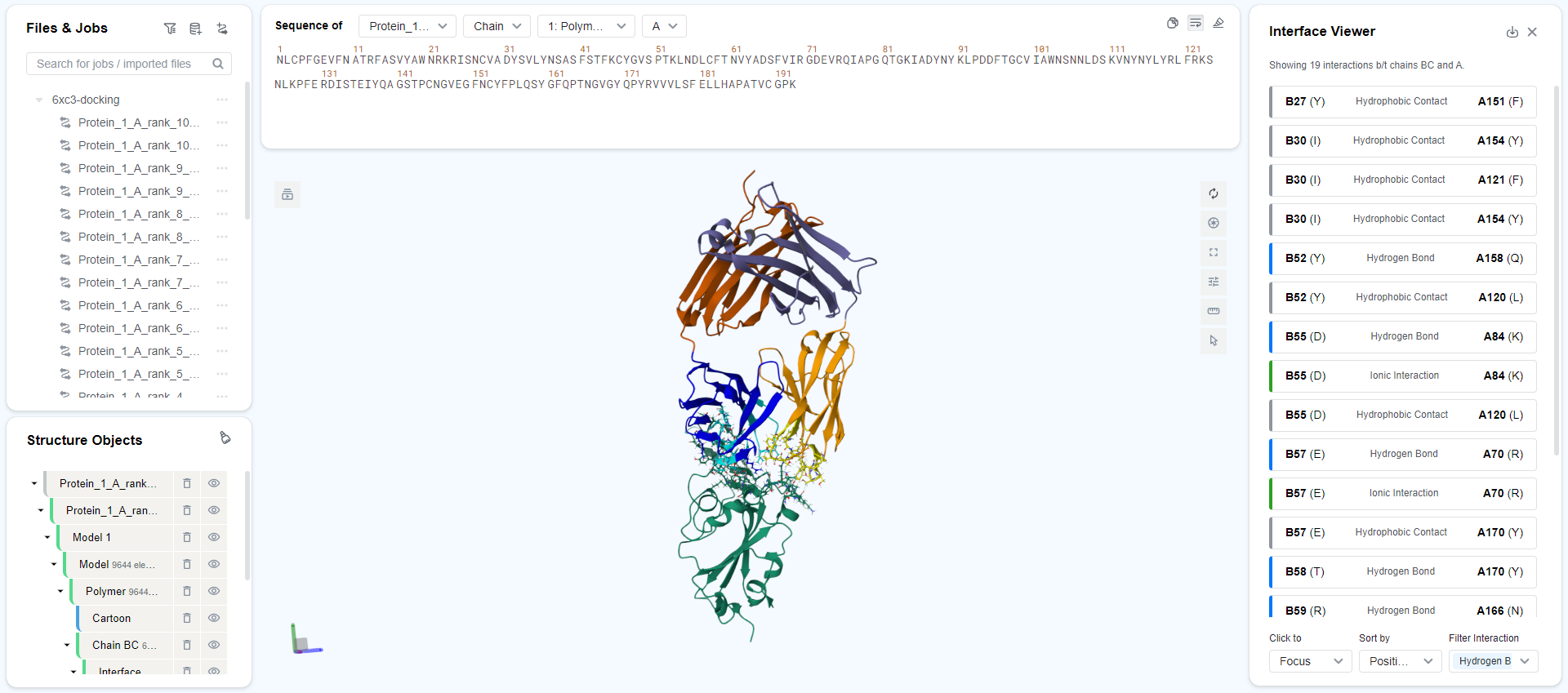
In the Ops column on the right, you can click the "![]() " button to view the complex structure. This will automatically redirect to the Mol* Viewer to examine the specific structure and docking details of the antibody-antigen complex.
" button to view the complex structure. This will automatically redirect to the Mol* Viewer to examine the specific structure and docking details of the antibody-antigen complex.
The antigens in all predicted structures are pre-aligned for you to compare the differences in antibody positions. If you need to align predicted and real complex structures, refer to the Structure Alignment documentation.
Next steps¶
Based on the predicted antibody-antigen complex structure, you might want to perform structure-based affinity maturation to optimize the binding affinity of your antibody-antigen complex.