Structure Alignment¶
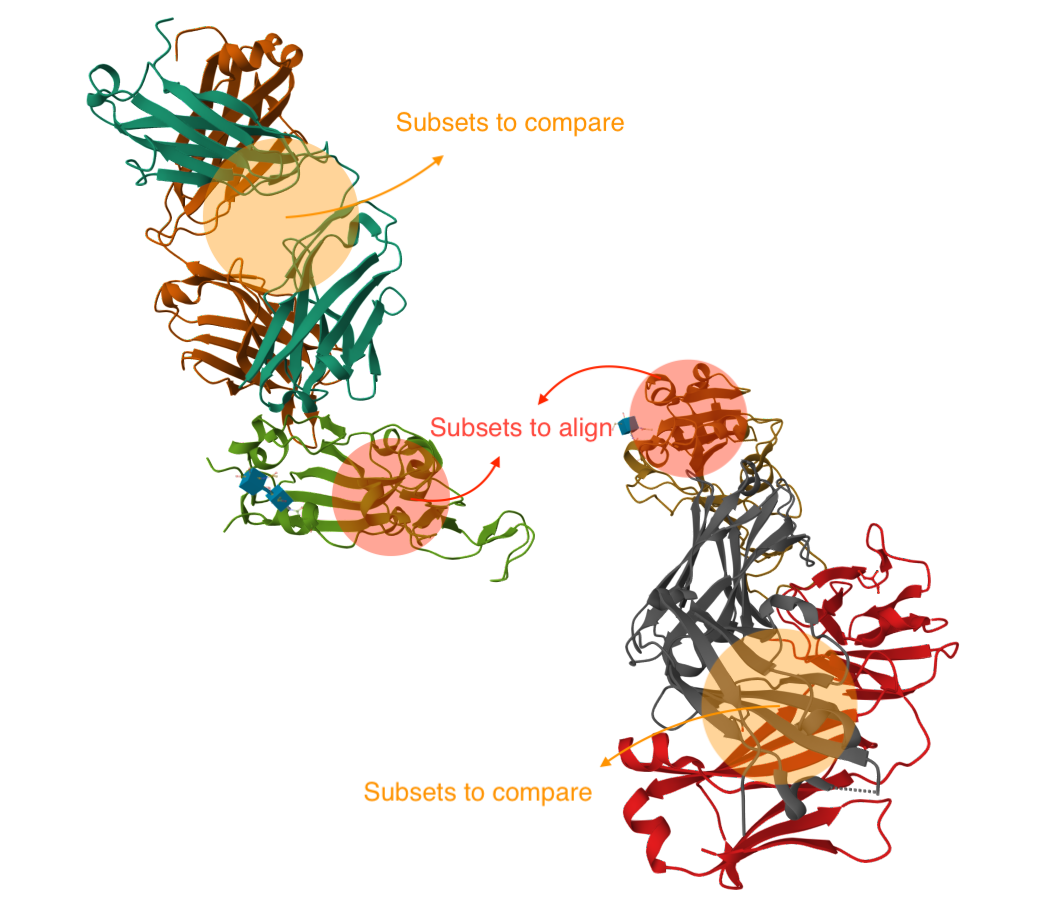
Structure align is a task of aligning a set of mobile structures to a targeted structure based on specified subsets. You could optionally compare another specified subset (e.g. calculate the ligand RMSD) after the alignment.
Inputs¶
To submit a Structure Align job, open the Project Editor and click "New Job" button on the left sidebar. Then click "Structure Alignment" under the "Utils" group to open the job submission page.
- Mobile Structures: Structures to be superimposed to the reference. You may select any structure files within your project. You may conveniently select all the output files of a job as the input.
- Reference Structures: A single structure serving as the reference for alignment. You may select any structure file within your project.
-
Subset to Align: A pair of substructures dictating the parts to align.
- Enter the chains and (optionally) residue numbers in the mobile and reference structures that you want to align. You may leave the residue number empty if you want to align the whole chain.
Requirements for Chains/Residues
- The residue numbers is a comma-separated list of individual residue numbers or residue number segments. E.g., "1,5-10,16".
- The specified mobile chains/residues must be present in all mobile structures. Otherwise, an error will be raised.
- The specified reference chains/residues must be present in the reference structure. Otherwise, an error will be raised.
- To add a chain to the substructure to align, click on the "
 " button. To remove a chain, click on the "
" button. To remove a chain, click on the " " button.
" button.
Selecting Residues in the Structure Viewer
You don't have to manually enter the residue numbers. After you click on the residues input field, you can click on the "open file" text below the input field to open the corresponding structure in the Structure Viewer. After selecting the residues in the Structure Viewer, click "import from selection" to fill in the input fields with the selected residues.
-
Subsets to Compare (optional): A pair of substructures specifying the parts to compare (e.g. calculate the ligand RMSD) after the alignment. This input is optional unless the model is set to DockQ.
- Job Name: Name of the job. Note that the job name must be unique within the project.
Model & Parameters¶
- Align: Align works well for proteins with decent sequence similarity (identity >30%).
- Super: Super works well for proteins with high structure similarity but low sequence similarity (it is sequence-independent).
- CEAlign: CEAlign works well for proteins with low sequence similarity but is much slower. Uses the combinatorial extension (CE) algorithm to align two proteins.
- DockQ: DockQ is a continuous quality measure for protein docking models based on the CAPRI evaluation protocol.

Results¶
Click the job name in the Files & Jobs panel to view the job results.
The results are stored in a CSV file, which can be downloaded by clicking the "![]() " button to the top-right of the Summary Table.
" button to the top-right of the Summary Table.
The summary table contains the following columns if the model is not "DockQ". If subsets to compare are not specified, the optional columns will be omitted.
- Input (str): Name of the mobile file.
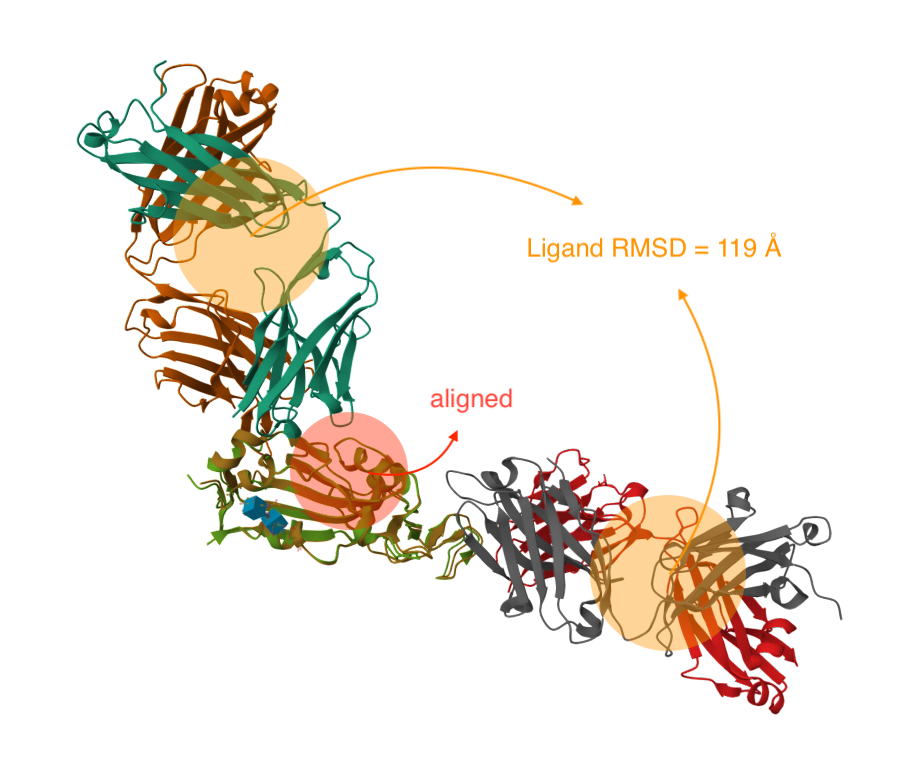
- RMSD (float): Root mean square deviation between the mobile and reference subsets to align after alignment and refinement (rejection of structural outliers).
- Raw RMSD (float): Raw root mean square deviation between the mobile and reference subsets to align without rejection of structural outliers.
- #res (aln) (int): Number of residues that remain in the mobile subset to align after refinement.
- #res (tot) (int): Total number of residues in the mobile subset to align.
- Ligand RMSD (float, optional): Root mean square deviation between the the mobile and reference subsets to compare without rejection of structural outliers.
- Ligand #res (aln) (int, optional): Number of residues that remain in the mobile subset to compare after refinement.
- Ligand #res (tot) (int, optional): Total number of ligand residues in the mobile subset to compare.
- Aln. mat. (4x4 float array): 4x4 transformation matrix for the alignment.
- Seq. align file (str): Name of the output sequence alignment file (.aln).
- Output file (str): Name of the output aligned mobile file (.pdb).
Rejection of Structural Outliers
The alignment algorithm may reject some outlier residues (> 2Å RMS) during the alignment process to make the rest of the alignment more robust. The raw/final RMSD values are calculated before/after removing these outliers.
Alignment Matrix
The alignment matrix is a 4x4 transformation matrix that can be used to transform the mobile structure to the aligned state. The matrix is defined as:
For each atom with position \(\boldsymbol{x}\) in the mobile structure, the position of the same atom in the transformed (aligned) mobile structure \(\boldsymbol{x}'\) is calculated as:
The summary table contains the following columns if the model is "DockQ".
- Input (str): Name of the mobile file.
- fnat (float): the fraction of mobile contacts in the native (reference) structure (TP/T). Higher is better.
- fnonnat (float): the fraction of non-native (not-in-reference) contacts in the mobile structure (FP/P). Lower is better.
- iRMS (float): the RMSD between the contacts (interface residues) of the mobile and reference structures.
- lRMS (float): the RMSD between the ligand (subsets to compare) residues of the mobile and reference structures after aligning the receptor (subsets to align).
- DockQ (float): the DockQ score.
- quality (str): the docking quality of the mobile structure, one of 'Incorrect', 'Acceptable', 'Medium', or 'High'.
- Interface aln mat (4x4 float array): a (4, 4) transformation matrix for the interface alignment.
- Receptor aln mat (4x4 float array): a (4, 4) transformation matrix for the receptor (subsets to align) alignment.
- Interface-aligned file (StrPath): Name of the interface-aligned mobile structure.
- Receptor-aligned file (StrPath): Name of the receptor-aligned mobile structure.
DockQ Score
The DockQ score is a continuous quality measure for protein docking models based on the CAPRI evaluation protocol. It ranges from 0 to 1, with higher values indicating better quality. The score is defined as:
In DockQ calculations, the following terms are equivalent.
| In this job | subsets to align | subsets to compare |
|---|---|---|
| In protein-protein docking | receptor | ligand |
| In antibody-antigen docking | antigen | antibody |
The interface residues, or contacts, are defined as residues in a subset with at least one heavy atom within 5 Å of any heavy atom in the other subset.