抗原特异 CDR 库的从头设计¶
抗原特异 CDR 库的从头设计旨在利用 AI 从头设计针对给定抗原表位的抗体 CDR 库。这个功能在针对一些难成药表位(通过动物免疫等传统手段难以获得高质量的结合抗体)时尤为有用。
挑战¶
CDR 库设计的搜索空间非常庞大,我们需要基于可能的抗体-抗体及抗原-抗体相互作用,确定十多个残基的氨基酸类型和位置。传统方法难以在计算的效率和准确性之间取得平衡。然而,生成式 AI 和几何深度学习技术的突破使得抗体设计的成功率达到了全新的水平。
任务原理¶
为帮助您更好地理解任务的输入、输出,实现更好的设计结果,本节将对任务的原理进行简要介绍。
- 从头设计序列和结构: 我们自研的生成模型会设计能结合到您指定表位的抗体 CDR 区的序列和结构。
- 更多序列设计: 为了进一步增加序列多样性,我们会基于上一步得到的抗原-抗体复合物结构,生成更多 CDR 区的序列。
- 虚拟筛选: 为识别最有潜力的候选分子,我们会对生成的所有序列进行虚拟筛选。筛选条件可以在结果页面中更改。
输入¶
要提交抗原特异 CDR 库设计任务,请打开项目编辑器 并点击左侧边栏中的"New Job(新建任务)"按钮,然后选择"Antibody Design(抗体设计)"任务组中的"Targeted CDR Library Design(抗原特异 CDR 库设计)"。任务提交表单将在新标签页中打开。
-
Antigen(抗原): 目标抗原。
- Structure(结构): 选择一个包含目标抗原结构的 PDB 或 mmCIF 文件。如果该文件已经在结构查看器中打开,请点击 "Select from viewer"。如果该文件在当前项目中,请点击 "Select from project"。否则,您可以在文件与任务管理器中上传或从云数据库中导入。
- Chain(链): 指定抗原的链 ID,例如 "A,B"。
为多链抗原设计抗体
我们建议您只选择一条抗原链,因为当前模型可能会改变多个抗原链之间的相对位置。我们会在后续版本中修复这个问题。
-
Epitope(表位): 目标表位(抗体结合位点)所包含的氨基酸。输入以逗号分隔的连续序列片段,格式为
{chain}:{start_res}-{end_res},例如A:2-10,A:15-30,B:40-100。更多信息请参见表位格式。您也可以在结构查看器中选择表位,然后点击 "import from selection" 自动填充此输入框。从选择中导入表位
点击 "Epitope" 输入框,您会看到输入框下方有 "open file" 或 "import from selection"。
表位氨基酸的选择与结构多样性
请注意,您需要输入的不是待设计的几个 CDR 残基的结合位点,而是整个抗体的结合位点。
较小的表位输入往往会导致较低的结构多样性。如果您希望设计一个结构多样性较高的库,请尝试输入至少6个表位氨基酸。 然而,表位输入过大可能会导致设计成功率偏低,因为模型会试图设计能结合绝大部分表位氨基酸的抗体,因而产生不切实际的设计。一个解决方案是启动多个目标表位不同的设计任务。
-
Antibody(抗体): 待设计的抗体。
- Antibody Name(名称): 抗体的名称。默认为“Antibody”。要更改名称,请将鼠标悬停在抗体名称上,然后点击 "
 " 按钮。
" 按钮。 - Format(格式): 您可在下拉菜单中切换抗体的格式。可选项包括“VH+HL”(正常抗体,默认)和“VH”(纳米抗体)。
- Sequence(序列): 抗体模板序列。待设计的 CDR 区应替换为占位符
[{region}, {init_seq}]。以下是一个示例。
指定待设计的 CDR 区
{region}标签指明了待设计的 CDR 区(H1/H2/H3/L1/L2/L3)。- H1/2/3 标签只能出现在重链,L1/2/3 标签只能出现在轻链。
- 每个 CDR 区标签只能出现一次,例如,输入中不能出现两个 "[H3, ...]" 标签。
- 当前版本中,一次只能设计一条链上的 CDR 区。例如,您可以同时设计 H1、H2 和 H3(都在重链上),但不能同时设计 H3 和 L3。请注意,待设计的 CDR 区氨基酸较多时,设计成功率可能会降低。
{init_seq}是待设计 CDR 区的初始序列,其中 X 表示待设计的残基。- 例如,
[H1, XXGSX]表示在 H1 区设计 3 个残基,而氨基酸 "GS" 是固定不变的,会在所有生成的序列中出现。 - 如果待设计 CDR 区的所有残基都需要设计,只需在
{init_seq}中输入 CDR 区的长度即可,例如[H3, 11]表示设计 H3 区的 11 个残基。 - 当前版本中,每个任务只能生成长度相同的抗体。如果您需要设计由不同长度的抗体组成的文库时,请启动多个任务。
 (上传序列): 点击 "" 按钮上传您自己的抗体模板(一个 .FASTA 文件)。如果格式为“VH+VL”,您的 FASTA 文件中必须有两条链,且标签必须具有相同的前缀且分别以 "_H" 和 "_L" 结尾,如上所示。如果格式为“VH”,您的 FASTA 文件中必须只有一条链,且标签必须以 "_H" 结尾。
(上传序列): 点击 "" 按钮上传您自己的抗体模板(一个 .FASTA 文件)。如果格式为“VH+VL”,您的 FASTA 文件中必须有两条链,且标签必须具有相同的前缀且分别以 "_H" 和 "_L" 结尾,如上所示。如果格式为“VH”,您的 FASTA 文件中必须只有一条链,且标签必须以 "_H" 结尾。- Job Name(任务名称): 任务的名称。请注意,任务名称必须在项目内唯一。
- Antibody Name(名称): 抗体的名称。默认为“Antibody”。要更改名称,请将鼠标悬停在抗体名称上,然后点击 "

表位格式¶
GeoBiologics 中表位氨基酸的列表由一个或多个“位点”和“片段”组成。
- 每个“位点”表示为
{chain_id}:{res_id},例如H:100。 - 每个“片段”表示为
{chain_id}:{start_res_id}-{end_res_id}。例如H:100-112。注意,片段起止氨基酸编号不能携带插入码,以免带来歧义。例如,某些抗体可能存在这样的 IMGT 编号:"110 111 111A 112B 112A 112",此时如果您指定H:110-112,我们将难以判断您具体需要突变哪些氨基酸,所以这种情况下,建议您使用位点表示。
多个位点或片段以半角(英文)逗号或换行符隔开,例如H:100-110,H:111,H:111A,H:112B,L:50-60,L:92。
模型 & 参数¶
您可使用我们自研的生成模型 GeoFlow-Design 进行高质量的抗体设计。模型参数如下:
- # designs: 生成的结构设计的数量(默认 100)。请注意,序列设计的数量约为结构设计数量的三倍(具体数量会在任务小结中展示),实际通过虚拟筛选的序列数通常小于此数量。因此,在实际项目中,建议您先尝试运行 200 个设计,然后根据设计和筛选的成功率调整设计数量。
- Temperature: 序列采样温度(默认 1)。较低的温度会导致更丰富的结构多样性。推荐在 0.8 到 1.2 之间。
- seed: 结构初始化随机种子(默认随机整数)。
结果¶
创建任务后,您可以点击文件与任务管理器面板中的作业名称查看结果。
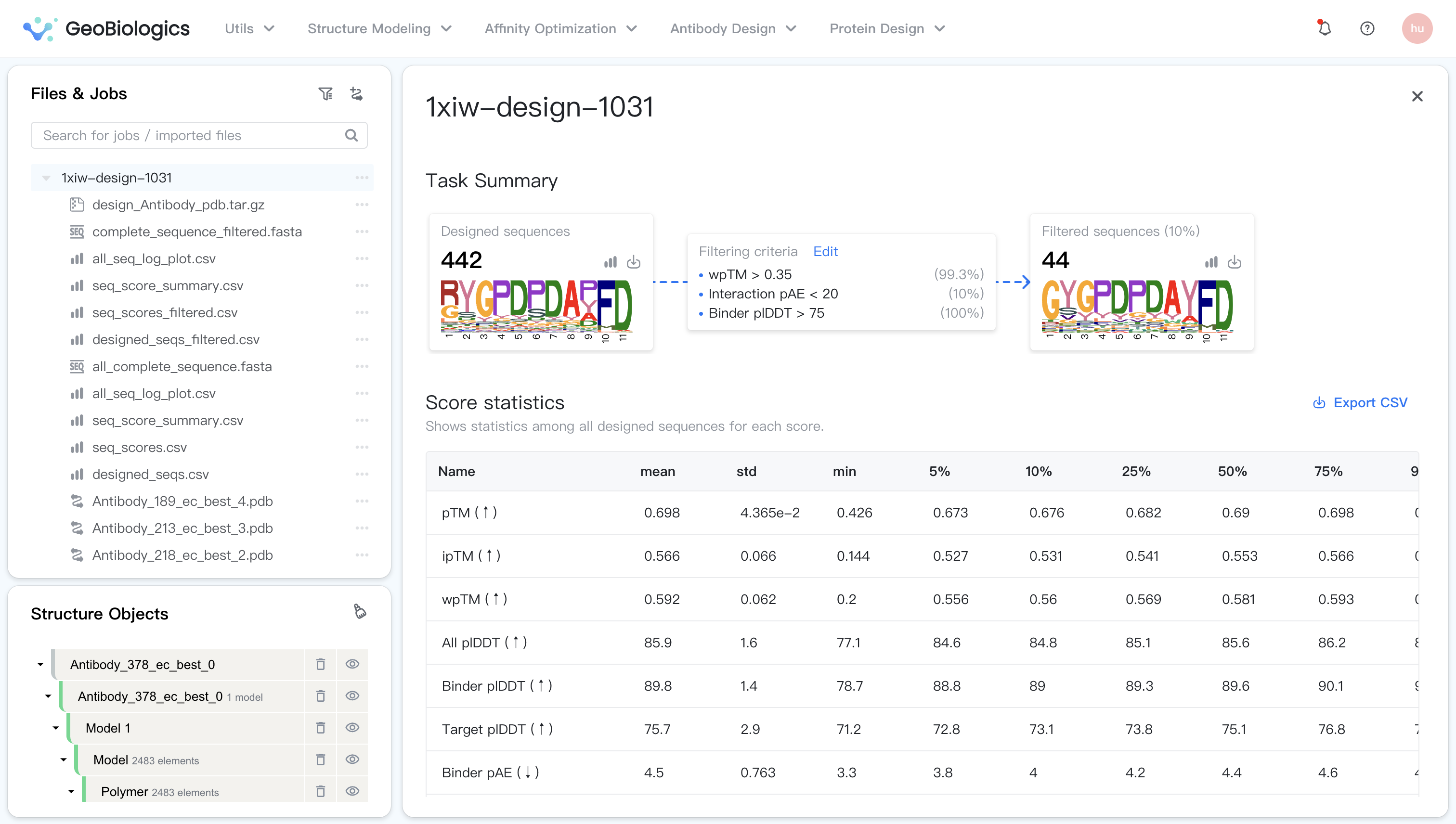
任务小结¶
任务小结展示了设计后序列文库、虚拟筛选条件、筛选后序列文库的基本信息。
- Designed sequences(设计后的序列文库): 粗体数字显示了设计后序列文库的大小。
 : 点击查看前 500 个设计的序列及其打分。
: 点击查看前 500 个设计的序列及其打分。 : 鼠标悬浮后会弹出悬浮菜单,您可点击下载(1)AI 模型设计的序列及其打分;(2)设计出的完整抗体序列;(3)所有设计的复合物结构。
: 鼠标悬浮后会弹出悬浮菜单,您可点击下载(1)AI 模型设计的序列及其打分;(2)设计出的完整抗体序列;(3)所有设计的复合物结构。- Logo 图:下方的 logo 图绘为您展示设计出的所有序列的氨基酸分布。
- Filtered sequences(筛选后的序列文库): 粗体数字显示了筛选后序列文库的大小。
- : 点击查看前 500 个筛选后的序列及其打分。
- : 鼠标悬浮后会弹出悬浮菜单,您可点击下载(1)AI 模型筛选后的序列及其打分;(2)完整筛选的序列。
- Filtering criteria(筛选条件): 虚拟筛选的筛选条件。每个条件旁边会用灰色字显示通过筛选的序列所占的比例。您可以点击“Edit”来更改筛选条件,筛选后的序列文库会随之更新。
虚拟筛选中使用的打分项
虚拟筛选环节,我们会使用自研的 GeoFlow 模型评估设计出来的复合物结构与序列之间的一致性(consistency),或者说匹配度。具体打分指标包括:
- pTM: 预测的 TM (Template modeling) 分数。分数越高,表示复合物结构的整体置信度越高。注意,这个分数是整个复合物的平均值,所以容易被某个大的结构域主导。
- ipTM: 预测的复合物接口区(antibody-antigen interface)的 TM 分数。分数越高,表示复合物结合界面的置信度越高。
- wpTM: 预测的复合物加权 TM 分数,计算公式为 \(\mathrm{wpTM} = 0.8 \times \mathrm{ipTM} + 0.2 \times \mathrm{pTM}\)。这个分数综合考虑了复合物整体结构和结合界面的置信度,是一个比较可靠的打分指标。
- All plDDT: 预测的复合物所有残基的 lDDT (local Distance Difference Test) 分数的平均值。分数越高,表示复合物整体序列-结构一致性越高。
- Binder plDDT: 预测的复合物抗体部分的 lDDT 分数的平均值。分数越高,表示抗体序列-结构一致性越高。
- Target plDDT: 预测的复合物抗原部分的 lDDT 分数的平均值。分数越高,表示抗原序列-结构一致性越高。
- Binder pAE: 预测的复合物抗体部分的 aligned error (AE) 的平均值。分数越低,表示抗体序列-结构一致性越高。
- Target pAE: 预测的复合物抗原部分的 aligned error (AE) 的平均值。分数越低,表示抗原序列-结构一致性越高。
- Interaction pAE: 预测的复合物抗体-抗原相互作用部分的 aligned error (AE) 的平均值。分数越低,表示抗体-抗原相互作用的一致性越高。
- Binder-aligned lRMSD: 预测与设计的复合物结构之间,抗体的均方根偏差(RMSD)。分数越低,表示抗体的序列-结构一致性越高。
- Target-aligned lRMSD: 预测与设计的复合物结构之间,抗原对齐后,抗体的均方根偏差(RMSD)。分数越低,表示表位的序列-结构一致性越高。该分数很高时,表示预测的表位与设计的表位不一致。
打分的统计量¶
本节用表格展示了各打分指标在所有设计序列上的统计量,包括均值、标准差、最小值、最大值,以及 5%、10%、25%、50%、75%、90% 和 95% 百分位数。您可以基于这些统计量来理解打分的分布,并设置筛选条件。
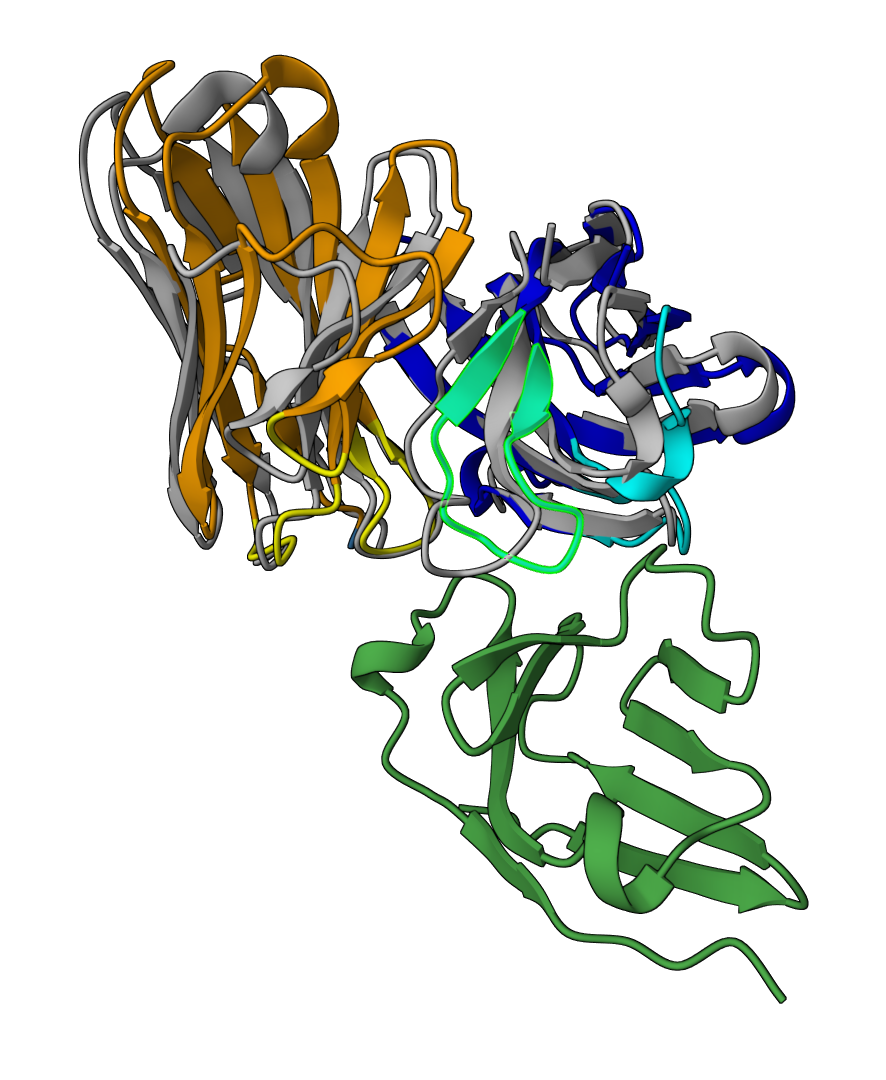
代表结构¶
为了帮助您方便地查看设计的复合物结构,我们对设计出的所有结构进行了聚类,并展示每个聚类中的代表结构。 请注意,本节表格中的 Epitope residues(表位氨基酸)是按照输出结构中的编号进行编号的,这些编号与输入结构中的编号可能不一致。 您可以点击 "open" 在结构查看器中打开这些结构。