蛋白结构预测¶
蛋白结构预测是一项根据氨基酸序列预测蛋白三维结构的任务。由于结构决定功能,蛋白结构预测在生物学和医学领域具有至关重要的作用。在这里,我们提供两种先进的蛋白结构预测方法:AlphaFold2-Multimer v3 和 Protenix。
功能亮点¶
-
全原子建模: 在GeoBiologics上,您可以预测几乎所有生物分子——包括蛋白、RNA、DNA、小分子及其复合物的结构。
-
批量预测: 支持在云端对多条序列进行批量预测,同时可以选择使用Amber松弛法(可选)。
-
图形界面: 无需配置环境或使用命令行;仅需在图形界面中点击即可完成结构预测。
Protenix¶
输入¶
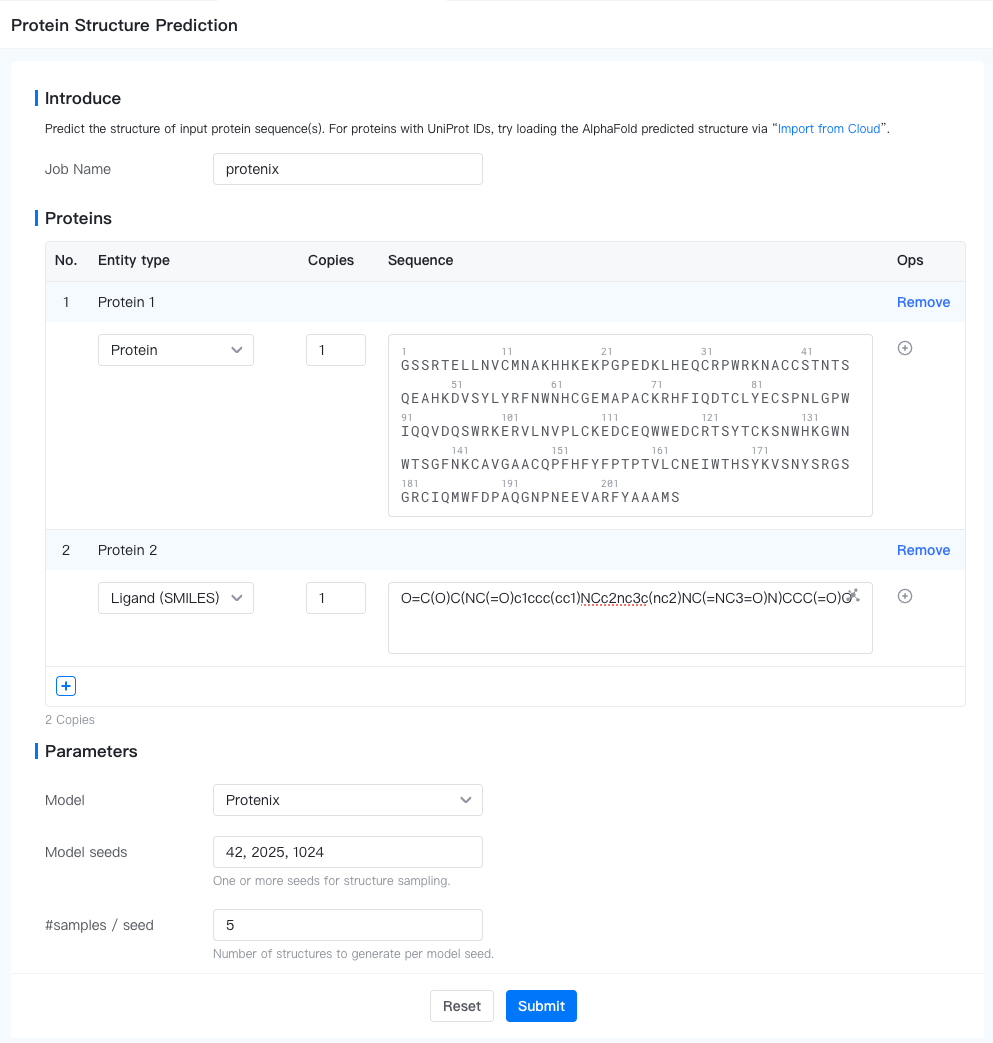
要提交蛋白结构预测任务,请打开项目编辑器并点击左侧边栏中的"New Job(新建任务)"按钮,然后选择"Structure Modeling(结构建模)"任务组中的"Protein Structure Prediction(蛋白结构预测)"。任务提交表单将在新标签页中打开。
-
Biomolecules(生物分子): 描述您的生物分子。您可以在一个任务中预测多个生物分子,每个生物分子将被独立预测。每个生物分子内可以包含多个实体。
 : 在输入中添加新的生物分子。
: 在输入中添加新的生物分子。- Biomolecule Name(生物分子名称): 生物分子的名称。默认为"蛋白 {i}"。要更改名称,请将鼠标悬停在生物分子名称上方,然后点击 "
 " 按钮。
" 按钮。 - Entity Type(实体类型): 生物分子中实体的类型。有效选择为 "蛋白", "RNA", "DNA", "配体 (SMILES)"。
- Copies(副本数): 当前实体的副本数。默认为 1。
- Sequence(序列): 当前实体的序列。不同实体类型有不同的序列格式:
- Protein(蛋白): 输入氨基酸序列,使用CCD编码,例如 (ACE)GQLEEIAKQLEEIAWQLEEIAQG(NH2)。
- RNA/DNA(RNA/DNA): 输入核苷酸序列,使用CCD编码,例如 GCGAGUAAU(8OG)UUAC。
- Ligand (SMILES)(配体 (SMILES)): 输入SMILES字符串,例如 CCCC(C=O)O。
- Add Entity(添加实体): 点击 "
 " 按钮以添加新的实体到生物分子。您可以添加最多6个实体到生物分子。
" 按钮以添加新的实体到生物分子。您可以添加最多6个实体到生物分子。
参数¶
- Model seeds(模型种子): 结构采样的种子。多个种子会增加采样到高质量结构的概率,但会增加计算成本。
- #samples/seed(每个种子样本数): 每个种子生成的样本数。
结果¶
在文件与任务管理器面板中点击任务结果,查看任务结果。
结果摘要存储在CSV文件中,可以通过点击结果表格右上角的 "![]() " 按钮下载。
" 按钮下载。
结果表格包含以下列:
- name(名称): 与输入中的生物分子名称相同。
- seed(种子): 用于结构采样的种子。
- sample(样本): 当前生物分子和种子下的样本索引。
- ranking_score(排名得分): 模型对当前样本的排名得分。数值越高则效果越好。
- gpDE(全局预测距离误差): 当前样本的全局预测距离误差。数值越低则效果越好。
在最右边的列中,您可以点击 "![]() " 按钮查看预测的结构。
" 按钮查看预测的结构。
AlphaFold2-Multimer v3¶
输入¶
要提交蛋白结构预测任务,请打开项目编辑器并点击左侧边栏中的"New Job(新建任务)"按钮,然后选择"Structure Modeling(结构建模)"任务组中的"Protein Structure Prediction(蛋白结构预测)"。任务提交表单将在新标签页中打开。
-
Proteins(蛋白): 以 FASTA 格式输入蛋白序列。您可以在输入框中输入序列,也可以上传 FASTA 文件 (点击 "
 " 按钮). 每个蛋白(可能是多链的)都会独立预测。
" 按钮). 每个蛋白(可能是多链的)都会独立预测。 : 在输入中添加新的蛋白序列。
: 在输入中添加新的蛋白序列。- Protein Name(蛋白名称): 蛋白的名称。默认为第 i 个序列的"蛋白 {i}"。要更改名称,请将鼠标悬停在蛋白名称上方,然后点击 "" 按钮.
- Chain ID(链ID): 标识蛋白中链的单个字符。默认为按字母顺序排列的大写字母。您可以在 "Chain
- Add Chain(添加链): 向蛋白中添加新的链。AF2-Multimer 可以预测多链蛋白的结构。我们建议链数不超过 9 条,总残基数不超过 1000。
-
Job Name(任务名称): 任务的名称。请注意,任务名称在项目中必须是唯一的。

参数¶
- # cycles(循环次数): 要运行的循环次数(1 - 48)。默认为 16。
- Relax structure(松弛结构): 是否使用Amber对模型生成的蛋白进行松弛(默认为 false)。
- MSA mode(MSA模式): 用 MMSeqs2 从 UniRef 序列数据库以及可选的环境序列中搜索MSA。有效选择有 "MMSeqs2 (UniRef + Environmental)"(默认),"MMSeqs2 (UniRef)", "Single sequence (No MSA)"。
- Pair mode(配对模式): 多链 MSA 设置。配对:使用来自同一物种的配对序列。非配对:对每个链使用单独的 MSA。有效选择为 "Unpaired", "Paired", "Unpaired + Paired"(默认)。
结果¶
在文件与任务管理器面板中点击任务结果,查看任务结果。
结果存储在CSV文件中,可以通过点击结果表格右上角的"![]() "按钮下载。
"按钮下载。
结果表格包含以下列:
- name(名称): 与输入中的 FASTA 标签相同。
- sequence(序列): 与输入中的 FASTA 序列相同。
- plddt: 为生成的蛋白预测的 5 个 AF2 模型的 lDDT(局部距离差异测试)评分。数值越高则效果越好。 每个残基的 lDDT 评分存储在输出 .pdb 文件的 b-factor 中。您可以通过将 Cartoon 表示法(Polymer 组件)的颜色主题更改为"原子属性>不确定性/无序"来查看它们。
- ptm: 为生成的蛋白预测的 5 个 AF2 模型的 TM(模板建模)评分。数值越高则效果越好。
在最右边的列中,您可以点击 "![]() " 按钮查看预测的结构。
如果您启用了"松弛"选项,您会在下拉菜单中找到两个文件:一个是未松弛的结构,另一个是松弛后的结构。
" 按钮查看预测的结构。
如果您启用了"松弛"选项,您会在下拉菜单中找到两个文件:一个是未松弛的结构,另一个是松弛后的结构。