结构对齐¶
结构对齐是将一组结构与目标结构按照指定的方式进行对齐的任务。在对齐后,您还可以对比结构中的另一部分(例如,计算配体 RMSD)。
输入¶
要提交结构对齐任务,请打开项目编辑器并从"Utils(辅助工具)"下拉菜单中选择"Structure Alignment(结构对齐)"。
- Mobile Structures(输入结构):需要对齐至到参考结构的蛋白。您可以选择项目中的一个或多个结构文件。在下拉菜单中,您可以方便地选择某个任务的全部输出文件作为此任务任务输入。
- Reference Structures(参考结构):对齐中作为参考的结构。您可以选择项目中的任意结构文件。
-
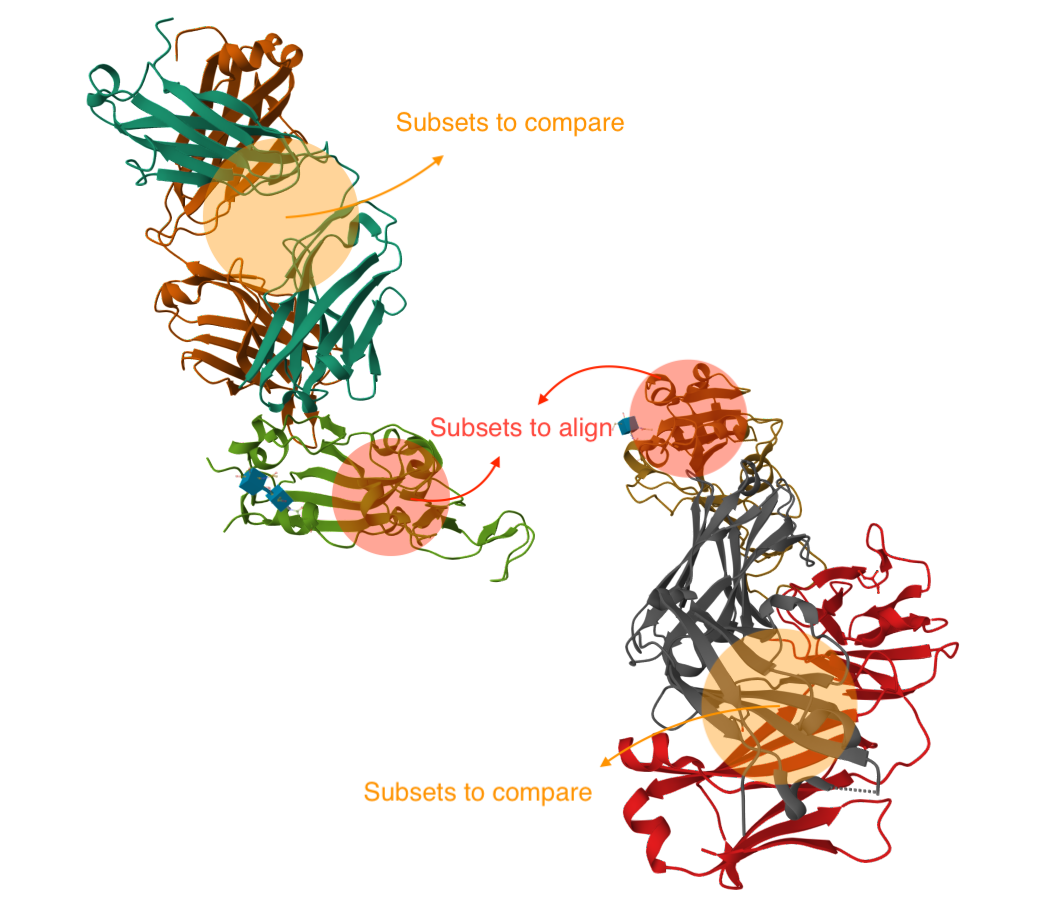
Subset to Align(待对齐子结构):待对齐的子结构。
- 填写您想要对齐的链和(可选)氨基酸编号来指定子结构。如果您想对齐整条链,则无需填写氨基酸编号。
链/氨基酸输入要求
- 在氨基酸编号输入框中,您需要输入一个由半角逗号 "," 分隔的单个氨基酸编号或氨基酸编号片段。例如,“1,5-10,16”。
- 您输入的输入结构中的链/氨基酸必须在所有输入结构中存在。否则将会有报错提示。
- 您输入的参考结构中的链/氨基酸必须在参考结构中存在。否则将会有报错提示。
- 如您想在待对齐子结构中添加一条链,请点击输入框后的“
 ”按钮。如您想删除一条链,请点击“
”按钮。如您想删除一条链,请点击“ ”按钮。
”按钮。
在结构查看器中选择氨基酸
您不必在输入框中手动输入氨基酸编号。 在您点击氨基酸编号输入框后,您可以点击输入框下方的“open file(打开文件)”文本以在结构查看器中打开相应的结构。在结构查看器中选中氨基酸后,点击“import from selection(从选区中导入)”即可自动填写输入框中的氨基酸。
-
Subsets to compare(待比较子结构,可选参数):对齐后需要进行比较(如计算配体 RMSD)的子结构。除非对齐的模型是 DockQ,否则此输入可不填。
- Job Name(任务名称): 任务的名称。请注意,任务名称必须在项目内唯一。
模型 & 参数¶
- Align:适用于序列相似性较高(大于 30%)的蛋白。
- Super:适用于结构相似性较高但序列相似性较低的蛋白(该方法与序列无关)。
- CEAlign:适用于序列相似性较低的蛋白,但速度很慢。使用组合扩展(CE)算法来对齐两个蛋白。
- DockQ:DockQ是基于 CAPRI 评估协议的值连续的质量评价指标,非常适用于蛋白对接模型的评估。

结果¶
在文件与任务管理器面板中点击任务结果,查看任务结果。
结果存储在 CSV 文件中,可以通过点击结果表格右上角的"![]() "按钮下载。
"按钮下载。
在您没有选择 DockQ 作为对齐模型时,结果表格包含以下列。如果未指定待比较子结构,下述可选列将不会出现在结果表格中。
- Input (str): 输入结构的文件名。
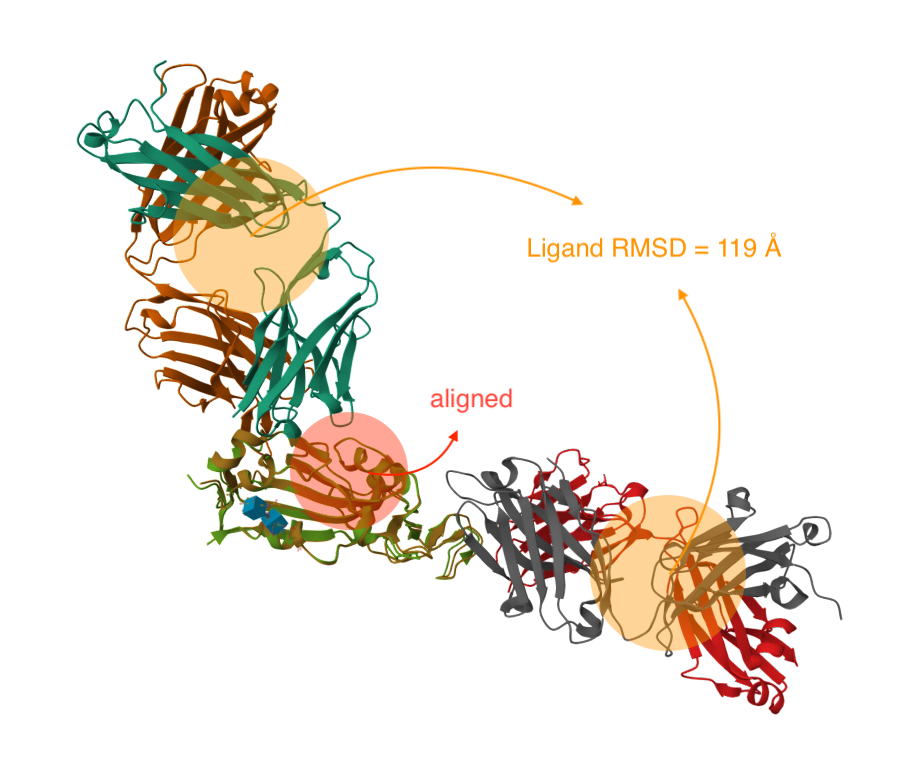
- RMSD (float): 对齐和精修(排除对不齐的异常氨基酸)后的输入结构和参考结构的待对齐子结构之间的均方根偏差。
- Raw RMSD (float): 对齐后、精修前,输入结构和参考结构的待对齐子结构之间的均方根偏差。
- #res (aln) (int): 精修后仍留在输入结构的待对齐子结构中的氨基酸个数。
- #res (tot) (int): 输入结构待对齐子结构中的氨基酸总数。
- Ligand RMSD (float, 可选): 对齐后、精修前,输入结构和参考结构的待比较子结构之间的均方根偏差。
- Ligand #res (aln) (int, 可选): 精修后仍留在输入结构的待比较子结构中的氨基酸个数。
- Ligand #res (tot) (int, 可选): 输入结构待比较子结构中的氨基酸总数。
- Aln. mat. (4x4 float array): 4x4 变换矩阵,用于变换输入结构,使其与参考结构对齐。
- Seq. align file (str): 任务输出,序列对齐文件(.aln)的文件名。
- Output file (str): 任务输出,对齐后的输入结构文件(.pdb)的文件名。
对齐精修与异常氨基酸的排除
在对齐过程中,对齐算法可能会排除一些对不齐的“异常”(RMS > 2Å)氨基酸。原始/最终 RMSD 值分别是在移除这些异常氨基酸之前/之后计算得到的。
对齐矩阵
对齐矩阵是一个 4x4 变换矩阵,可用于将输入结构变换到与参考结构对齐的状态。该矩阵定义如下:
对于输入结构中位置为 \(\boldsymbol{x}\) 的原子,变换(对齐)后的输入结构中相同原子的位置 \(\boldsymbol{x}'\) 计算如下:
在您选择 DockQ 作为对齐模型时,结果表格包含以下列。
- Input (str): 输入结构的文件名。
- fnat (float): 参考结构的界面氨基酸在输入结构中出现的比例(TP/T)。越高越好。
- fnonnat (float): 输入结构的界面氨基酸不在参考结构中出现的比例(FP/P)。越低越好。
- iRMS (float): 输入结构和参考结构的界面氨基酸之间的 RMSD。
- lRMS (float): 对齐后,输入结构和参考结构的待比较子结构(subsets to compare)之间的 RMSD,也称配体 RMSD。
- DockQ (float): DockQ 分数值。
- quality (str): 输入结构的对接质量,可能的取值为 'Incorrect', 'Acceptable', 'Medium', 或 'High'。
- Interface aln mat (4x4 float array): 4x4 变换矩阵,用于变换输入结构,使其界面与参考结构的界面对齐。
- Receptor aln mat (4x4 float array): 4x4 变换矩阵,用于变换输入结构,使其待对齐子结构(受体)与参考结构的待对齐子结构(受体)对齐。
- Interface-aligned file (StrPath): 界面对齐后的输入文件的文件路径。
- Receptor-aligned file (StrPath): 待对齐子结构(受体)对齐后的输入文件的文件路径。
DockQ 分数
DockQ 分数是基于 CAPRI 评估协议的蛋白对接模型的连续质量评价指标。分数范围为 0 到 1,数值越高表示质量越好。DockQ 分数定义如下:
在 DockQ 计算中,以下术语是等价的。
| 本任务中 | 待对齐子结构 | 待比较子结构 |
|---|---|---|
| 蛋白-蛋白对接中 | 受体 | 配体 |
| 抗体-抗原对接中 | 抗原 | 抗体 |
在 DockQ 计算中,界面氨基酸被定义为一个子集中至少有一个重原子与另一子集任意重原子之间的距离小于 5 Å 的氨基酸。