突变助手¶
突变助手是一个智能工具,可提供基于数据的突变建议来指导蛋白质工程工作。该功能可用于抗体优化、亲和力优化和蛋白优化任务。
概览¶
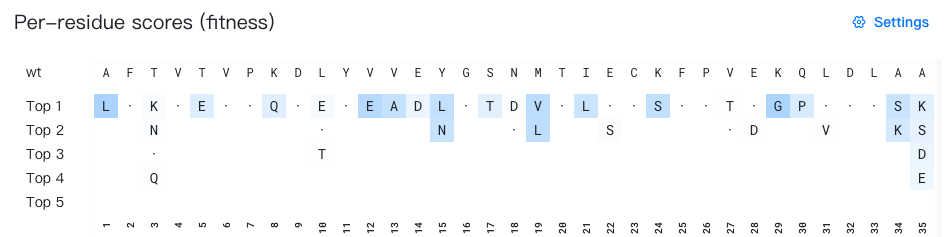
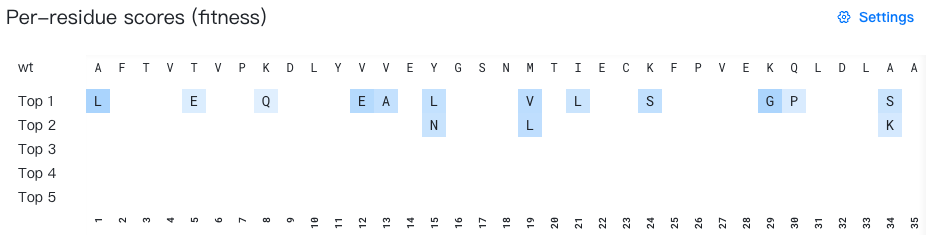
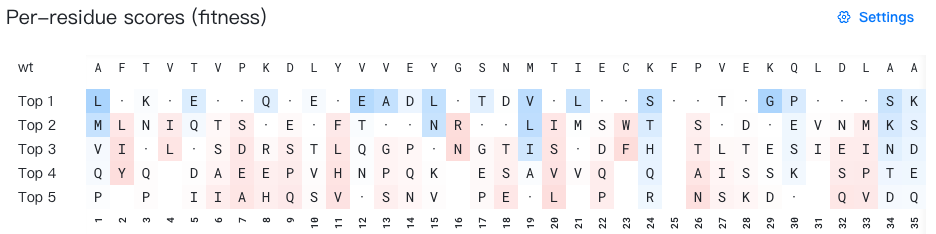
- 顶行:显示野生型序列(突变前的初始输入序列)。
- 底行:显示序列索引(从 1 开始编号)。
- 中间:显示每个位点的前 5 个突变建议。当野生型残基出现在前 5 个预测中时,显示为"·"以便于识别。所有残基根据所选的背景色数据源(Background color data source)着色。
什么是“每个位点的突变建议”?
位点 p 的第 k 个突变建议代表 AI 模型为该特定位点预测的第 k 个排名最高的残基。
例如,在上图中,为了生成位点 3 的突变建议,AI 模型会根据周围野生型序列的上下文评估哪些残基在该位点最有利。在上图中,位点 3 的前 4 个预测是 K、N、T 和 Q。由于 T 是该位点的野生型残基,因此显示为"·"。
重要提示:每一行代表单个位点的独立排名替代方案。单个"top-k"行中的残基不构成完整的序列变体。
显示设置¶

您可以点击 ![]() 按钮打开设置窗口,并配置以下"突变助手"选项:
按钮打开设置窗口,并配置以下"突变助手"选项:
背景色数据源¶
每个突变建议的背景颜色可以基于两种不同的指标:
- 原始分数(Raw score)(默认):每个位点上每个残基的原始预测分数。较高的正分数通常表示更好的性能,而负分数表示较差的性能。一个值得注意的例外是亲和力优化,其中更负的 \(\Delta\Delta G_\text{bind}\) 值代表更强的结合,因此是有利的。
- 位点内概率(Per-site probability):每个位点上每个残基的归一化概率,范围从 0 到 1。给定位点上所有 20 种可能氨基酸的概率总和为 1。由于概率值是按位点归一化的,因此比较不同位点的概率意义不大。
位点内概率如何计算?
位点内概率通过对每个位点的原始分数应用 softmax 函数来计算。
给定位点 \(p\) 上残基 \(i\) 的原始分数 \(s_{pi}\),位点 \(p\) 上残基 \(i\) 的位点内概率 \(p_{pi}\) 计算如下:
其中 \(T\) 是归一化温度(normalization temperature)——一个控制概率分布锐度的超参数。温度越高,概率分布越均匀;温度越低,高分和低分之间的差异越明显。
上述计算与统计力学中能量和概率的关系相似。
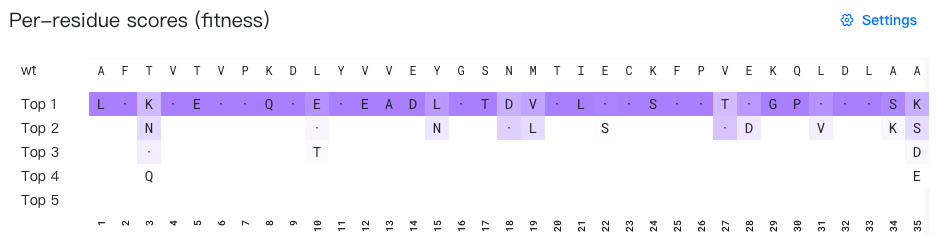
氨基酸背景色是如何确定的?
突变使用红-白-蓝渐变色标着色:
- 红色:预测表现比野生型差的突变(通常为负分数)
- 白色:中性突变(分数 ≈ 0)
- 蓝色:预测表现比野生型好的突变(通常为正分数)
正分数和负分数的颜色强度以不同的斜率缩放,即每单位分数的颜色变化率不同,以优化视觉辨别。
![]()
突变使用白色到紫色的渐变色标着色,颜色强度随位点内概率增加而增加。
![]()
显示条件¶
您可以使用三个独立的过滤器来自定义显示哪些突变:
-
原始分数排名(Rank of raw scores):按突变在所有位点的总体排名进行过滤。选项包括:
- "All"(全部)(默认):显示所有突变
- "Top 10"、"Top 20"、"Top 50"、"Top 100":仅显示在整个序列中按原始分数排名前 n 的突变
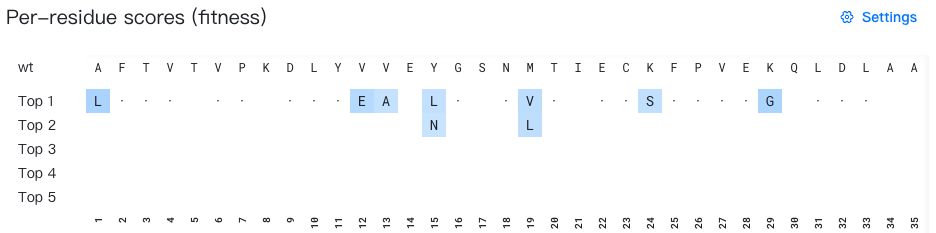
原始分数排名:Top 20 -
位点内概率范围(Per-site probability range):指定最小和最大位点内概率阈值。仅显示概率在此范围内的突变。默认范围是 0.01–1.0。
位点内概率范围:0-1 -
原始分数范围(Raw score range):指定最小和最大原始分数阈值。仅显示分数在此范围内的突变。默认范围包括所有突变(最小值到最大值)。
原始分数范围:1.0-5.3
Warning
设置限制性的概率或分数范围可能会从显示中过滤掉野生型残基。在极端条件下,可能会过滤掉所有突变。
归一化温度¶
归一化温度是一个控制概率分布锐度的超参数。温度越高,概率分布越均匀。温度越低,概率分布越尖锐。默认值为 0.125。
色阶范围¶
色阶范围定义了映射到渐变色中极端颜色的最小值和最大值:
- 对于原始分数模式:设置对应于最深红色(最小值)和最深蓝色(最大值)的分数值
- 对于位点内概率模式:设置对应于白色(最小值)和最深紫色(最大值)的概率值
您可以调整此范围以增强您感兴趣的值域的色彩对比度。超出指定范围的值将被显示为最近的极端颜色。